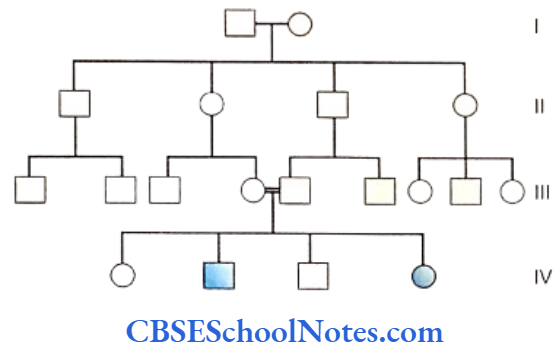
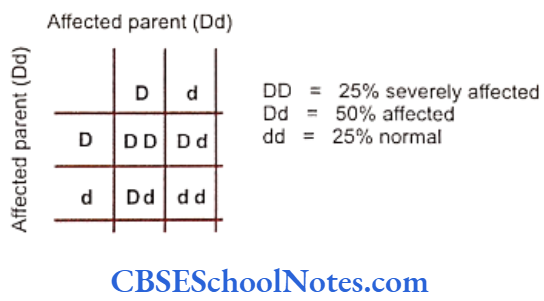
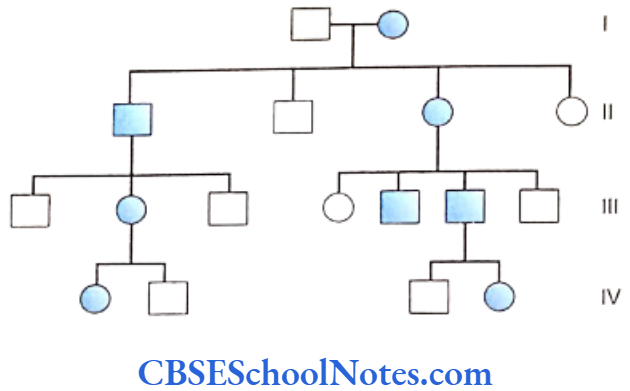
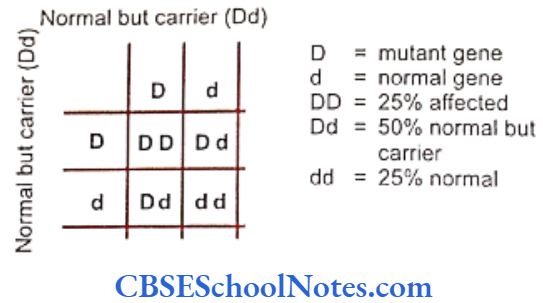
Genetics Of Developmental Disorders Of Teeth
Molecular (Genetic) Control Of Development Of Tooth
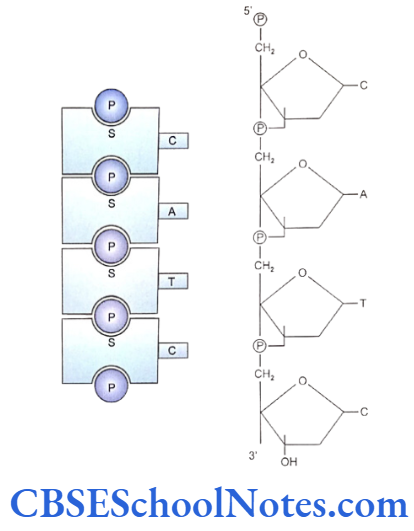
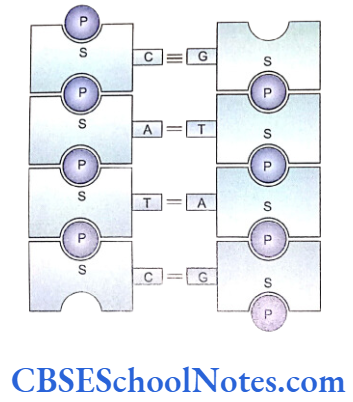
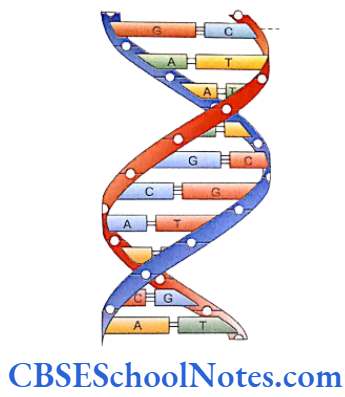
Each tooth has a distinct morphology and a specific location and determination of both the factors is under strict genetic control. Following description discusses the sequential molecular events that control the development of tooth and bring forth definite morphological stages during such development.
Our knowledge of molecular control of tooth development is mainly based on the studies conducted in mouse embryos. Most of the investigations and experiments on these embryos were carried out between embryonic age 9.0 and 17.0 days (E9.0- E17.0). These studies dealt with the expression of a number of transcription factors, signaling molecules, growth factor receptors and extracellular matrix molecules.
Teeth develop on the mandibular and maxillary arches through a series of interactions between the oral epithelium and underlying mesenchyme (i.e., the ectomesenchyme of first arch which is derived from the neural crest). Following are the main steps in the formation of a tooth.
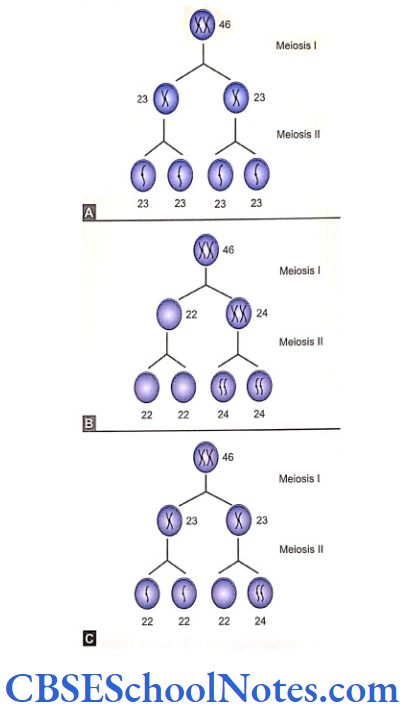
- A thickening of the oral epithelium appears at E11.5 in mice. This is the first morphological sign of tooth development. The thickened oral epithelium is now called as dental lamina.
- The dental lamina now grows into the underlying mesenchyme of the first branchial arch and forms the epithelial bud at E13.5. During the formation of epithelial bud the mesenchymal cells get condensed around the developing bud and form dental papilla. The dental papilla later forms tooth pulp and odontoblasts. The dentine is formed by these odontoblasts.
- On the embryonic day 14.5 (E14.5) the epithelial bud changes to a cap shaped structure. This stage is called as the cap stage. The cap undergoes further folding and forms a bell shaped structure (bell stage, E15.5). The epithelium of the bell eventually gives rise to ameloblasts, which form enamel.
- The central portion of the epithelium of cap forms a special signaling center called as enamel knot. The enamel knot is involved in the formation of tooth cusps.
Read and Learn More Genetics in Dentistry Notes
As stated earlier the formation of tooth begins with the interaction between the oral epithelium and the mesenchyme of the first branchial arch. It is believed that the oral epithelium of the first branchial arch sends signals to underlying mesenchyme. Under the influence of these signals the mesenchyme starts responding by expressing various regulatory genes.
How is the Oral-Aboral Axis Formed?
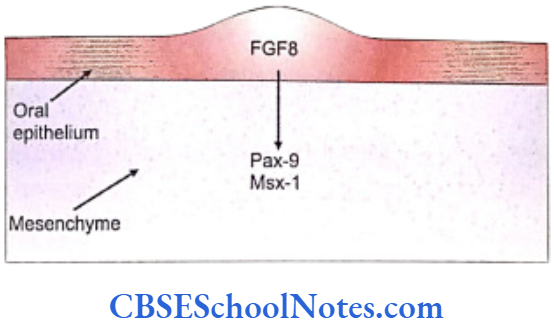
The oral epithelium of the first branchial arch from the day E9 starts expressing the gene Fgf-8 that secretes the fibroblast growth factor (FGF). This growth factor leads to the expression of Lhx-6 and Lhx-7 genes on the ectomesenchyme just beneath the oral epithelium.
Lhx genes are the Lim-homeobox domain genes which are expressed as transcription factors and control the pattern of tooth formation. The expression of Fgf-8 is restricted to the oral epithelium and matches very closely to the expression domain of Lhx-6/7 in the ectomesenchyme of the first branchial arch. The expression of these genes (Fgf-8, Lhx-6/7) thus establishes the oral-aboral axis.
How are the Sites of Formation of Tooth Germ Decided?
Many genes are involved in the formation of tooth germs. These are mentioned as under:
- The fibroblast growth factor encoded by the Fgf- 8 gene that acts on the underlying mesenchyme of the first branchial arch and induces the expression of Pax-9 gene (Carlson, 2004). The Pax-9 gene is the member of Pax gene family and encodes a paired domain containing transcription factor. This gene defines the localization of tooth germs. Thus the exact site of appearance of the tooth germ is decided by the expression of Pax-9 in the mesenchyme region at the prospective sites of all teeth.

- Pax-9 gene is expressed first in the prospective molar region (at E10) and then in the prospective incisor sites.
- The expression of Fgf-8 is widespread (not limited to only those areas where future tooth will form). However, the expression of Pax-9 is limited only to those sites, in the mesenchyme, where the future teeth germs would form. It means that there should be some mechanism by which the expression of Pax-9 is inhibited in mesenchyme where tooth is not destined to be formed.
- Both BMP-4 and BMP-2 molecules (Bone morphogenetic proteins) are expressed by Bmp- 4 and Bmp-2 genes. BMP-4 and BMP-2 proteins are able to inhibit the ‘Pax-9 inducing activity’ of Fgf-8 in the tooth mesenchyme. At quite early stages, Bmp-4 is expressed only in the epithelium but at later periods is expressed in mesenchyme also.
- The expression of Pax-9 is inhibited in those areas where teeth are not designed to develop.
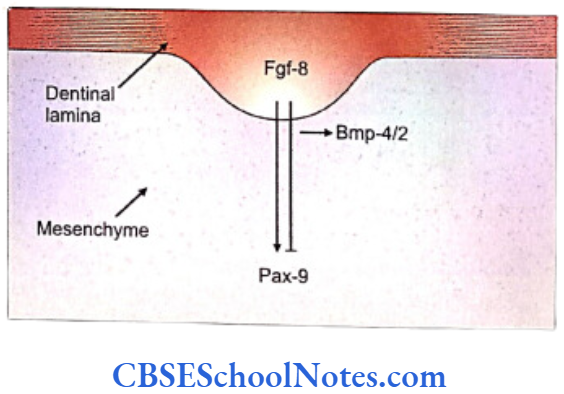
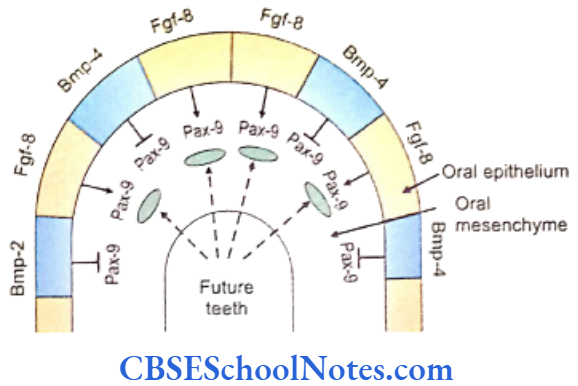
- Therefore the tooth germs develop only in those areas where Pax-9 expression is induced in the mesenchyme by the Fgf-8 expression in the overlying epithelium. Tooth germ does not develop in those areas where Bmp-4 signaling inhibits the ‘Pax-9 inducing activity’ of Fgf-8.
- Bmp-4 signaling also stimulates mesenchymal expression of Msx-1 and Bmp-4 itself. The Msx-1 (muscle specific homeobox like gene) expression is also induced by Fgf-8. Consequently, both Msx-1 and Pax-9 have similar functions i.e., both are essential in the mesenchyme for tooth morphogenesis. Thus, whereas Fgfs and Bmps have opposite effects on the expression of Pax-9, they both stimulate the expression of Msx-1.
- At E 11.5 the potential to direct the tooth development shifts from epithelium to mesenchyme. The expression of Bmp-4 also shifts now from the epithelium to mesenchyme. The Pax-9 and Msx-1 genes are co-expressed in the mesenchyme where the function of both the genes is required for the expression of Bmp-4.
- After E11.5 Pax-9 expression is not under the control of epithelium signals. Similarly, Bmps are no longer able to inhibit Pax-9 expression in mesenchyme.
- The Pax-9 and Msx-1 are essential for the establishment of the odontogenic potential of the mesenchyme.
- Lef-1 is first expressed in the dental epithelium (during dental lamina stage). Expression of this gene then shifts to mesenchyme during the bud stage. The mutation of Lef-1 gene (or Lef-1 knockout mice) shows the arrest of all dental development at its bud stage.
- The expression of Shh is localized in the dental epithelium at E11 and thus Shh is considered to be a good signaling candidate for tooth initiation. Shh stimulates epithelial cell proliferation at the sites of tooth development. Gli-2 and Gli-3 genes are two downstream mediators of Shh action. In case of Gli-2 and Gli-3 double mutation, embryos fail to produce any recognizable tooth bud.
How do Genes Control the Formation and Function of the Enamel Knot?
The enamel knot is the transient signaling center of the epithelium and directs the next phase of tooth development. The enamel knot is responsible for the formation of the tooth cusps that later give each individual teeth its characteristics surface.
- The development of the enamel knot is regulated by signals originating in the mesenchyme. One of the important functions of Pax-9 and Msx-1 is the maintenance of mesenchymal Bmp-4 expression. The Bmp-4 signaling is involved in the formation of the enamel knot.
- Bmp-4 induces the expression of p21, Bmp-2 and Msx-2 in the epithelium. These genes are associated with programmed cell death. The enamel knot is formed at the cap stage of dental development and secretes factors responsible for the apoptosis in the knot itself.
- At the same time the enamel knot secretes Fgf4 and Fgf9 which stimulate proliferation of certain neighboring cell compartments (enamel epithelium and dental papilla) (Fig. 10.4). As Fgf receptors are not present on the enamel knot cells, they do not respond to the Fgfs and thus fail to show proliferation.
- It is believed that Lef-1 gene (Hmg box containing transcription factor) mediates Fgf/BMP signaling in the epithelium at the late bud and early cap stage of tooth development. In the absence of Lef-1 tooth formation is arrested at the bud stage. The expression of Lef-1 has been shown to be inducible by Bmp-4. Thus Lef-1 is the mediator of Bmp-4 signaling in the epithelial tooth bud and is necessary for the establishment of enamel knot.
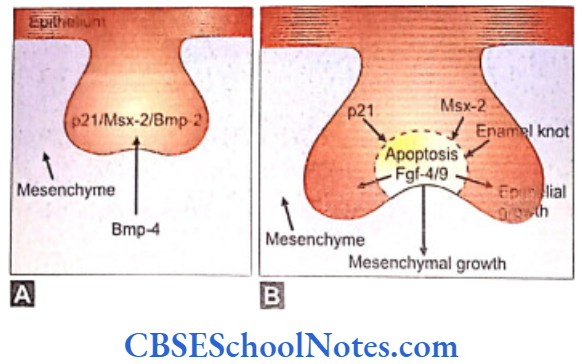
- Besides the above-mentioned genes, many other genes (i.e., Bmp-2, Bmp-7, Fgf-9, Slit-1 and Shh) are also expressed by cells of enamel knot. Shh expression is needed for the determination of bud morphology at E 13.
How are the Tooth Type and their Correct Position Determined?
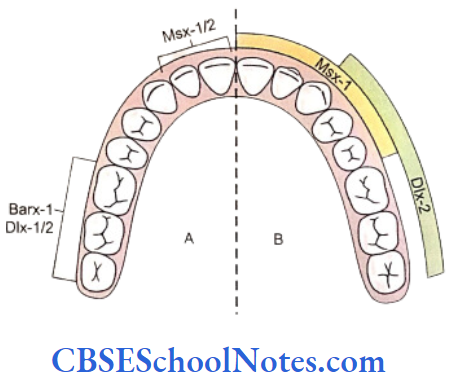
Most of the genes for dental patterning are expressed in the dental mesenchyme before E11. It has been suggested that there exists an odontogenic homeobox gene code that might determine the identity of each individual teeth. These homeobox genes include Msx-1, Msx-2, Dlx-1, 2, 3, 5, 6, and 7, Barx-1, Otlx-2, Lhx-6 and 7. These genes are expressed in specific spatial pattern in the mesenchyme of the first branchial arch which is derived from the neural crest cells of the mid-brain region.
- The instructions for tooth type formation are due to temporal regulation of homeobox genes expression in the ectomesenchyme, which is induced by ectodermal signals before E11.
- The release of Fgf-8 from the epithelium induces the expression of the homeobox gene Barx-1 in the mesenchyme of the proximal part (postrior part) of the first branchial arch. Presumptive molars develop in the proximal part of the first arch. Dlx-1 and Dlx-2 genes are also expressed in this region of the branchial arch and along with Barx-1 they are also involved in the formation of molar teeth. Though the Dlx-1 and Dlx-2 are expressed in both mandibular and maxillary arch but Dlx-5 and Dlx-6 are only expressed in mandibular arch. The mutation of both Dlx-1 and Dlx-2 leads to the absence of maxillary molars only. This is because the expression of Dlx-5/6 is responsible for the formation of mandibular molars. Thus Dlx-5/6 compensate for the loss of Dlx-1/2.
- The release of BMP-4 from the epithelium induces the expression of Msx genes in the distal part (anterior part) of the mesenchyme of first arch where presumptive incisor forms. Thus the expression of Msx-1 and Msx-2 in the distal mesenchyme is induced by BMP-4 and leads to the formation of the incisor teeth.
- It is believed that the formation of canine and premolars in human results due to overlapping domains of gene expression, i.e. overlapping expression of Msx and Dlx genes.
- The Msx genes are considered as incisor genes and Barx-1 as molar genes. The formation of canine and premolars in humans may be under the control of overlapping expression of Msx and Dlx genes.
Tooth Agenesis
Tooth agenesis is defined as deficiency in tooth number and it is one of the most common developmental anomalies in humans. The non- syndromic tooth agenesis is observed in various forms, i.e., partial or total (generalized). In mild form only one or few teeth are absent. While in its severe forms, many teeth are absent.
It should be noted that agenesis of third molars is much more common than the absence of other teeth (one or more third molars fail to develop in up to 35% of people in various population groups of the world). On the other hand the agenesis of deciduous teeth is quite rare; (below 1%) in various populations.
If we exclude the agenesis of the third molars, the absence of more than two teeth is observed in only about 1% of population. The absence of more than six teeth is very rare (0.1-0.3 % of population).
Tooth agenesis is usually an isolated anomaly (non- syndromic) in which tooth agenesis itself is the primary condition. However, it is also observed in association with oral clefts (Boogaard et al, 2000) and various malformation syndromes. When tooth agenesis is associated with syndromes it is usually severe in form.
Following is the list of syndromes and other congenital malformations in which tooth agenesis is found in conjunction with other developmental anomalies:
- Rieger’s syndrome
- Wolf-Hirschhorn syndrome • Williams syndrome
- Kabuki make up syndrome
- Cleft lip / cleft palate
- Ectodermal dysplasia
- Chondroectodermal dyspalsia
- Achondroplasia
- Holoprosencephaly.
The following description of tooth agenesis is confined to isolated or nonsyndromic agenesis.
Classification of Tooth Agenesis
Tooth agenesis is observed in following three forms:
Hypodontia refers to the developmental lack of a few teeth. The population frequency is over 5% (missing of wisdom teeth excluded).
Oligodontia refers to the developmental lack of more than six teeth (wisdom teeth not included). The population frequency is low especially for cases when the absence of teeth is the only malformation (“isolated” cases). Most often oligodontia appears as part of a congenital syndrome that affects several organ systems (ectodermal dysplasia, achondroplasia, chondroectodermal dysplasia, Rieger syndrome, etc.).
Anodontia refers to complete lack of teeth, which is very rare in occurrence.
Shapes and positions of existing teeth may also be abnormal in association with missing teeth. The features often seen include “peg-shaped” maxillary lateral incisors, “taurodontism” of molars and “mal- positions”.
Etiology of Tooth Agenesis
Though many environmental etiological factors for tooth agenesis have been identified, there is definitive proof that genetic factors play a major role in their etiology
Environmental factors that are implicated are: maternal systemic diseases (maternal diabetes, hypothyroidism, rubella infection during pregnancy), anticancer treatment during childhood (radiotherapy and chemotherapy).
Genetics of Tooth Agenesis
Pax-9 and Msx-1 are two key genes involved in the embryological development of teeth and their mutation leads to tooth agenesis (Mostowska et al., 2003: Matalova et al., 2008 and Kapadia, et al. 2007). Pax-9 is situated on chromosome 14 (14q21) and belongs to the Pax gene family that encodes a group of transcription factors playing a major role in early development.
Pax proteins are defined by the presence of a DNA-binding domain, the ‘paired domain’, which makes sequence-specific contact with the target DNA region. Msx-1 is a homeobox gene involved in numerous epithelial-mesenchymal interactions during vertebrate embryogenesis and appears to be incredibly significant during early tooth development. It is situated on the short arm of chromosome 4.
Pax-9 and Msx-1 encode transcription factors that are known to be essential for the switch in the odontogenic potential of developing tissues in the epithelium and the mesenchyme. These molecules play an important role in the maintenance of mesenchymal Bmp4 expression responsible for the formation of the dental organ.
Pax-9 is able to regulate Msx1 expression directly and interact with Msx-1 at the protein level to enhance its ability to transactivate Msx1 and Bmp4 expression during tooth development. Pax-9 and Msx1 act as partners in a signaling pathway that involves Bmp4.
Furthermore, the regulation of Bmp4 expression by the interaction of Pax-9 with Msx-1 at the level of transcription and through formation of a protein complex determines the fate of the transition from the bud to cap stage during tooth development.
Till date seven Msx-1 mutations as well as some whole gene deletions have been discovered in tooth agenesis patients. Msx-1 frameshift mutation is responsible for autosomal-dominant oligodontia without clefting or nail dysplasia. The mutation involves duplication of the guanine nucleotide at position 62 in exon 1 of the Msx-1 gene. This mutation in Msx1 is usually associated with the absence of multiple permanent teeth including all second bicuspids and mandibular central incisors.
A number of mutations (upto 15) have been identified in the Pax genes that include nonsense, missense, frameshift and deletion types of defects. Mutation in the initiation codon of Pax-9 causes severe or complete inhibition of Pax9 translation at one allele resulting in a reduced amount of Pax-9 transcription factor, representing a haploinsufficiency for Pax-9. This functional insufficiency or absence of Pax-9 protein produced from Pax-9 gene ultimately results in tooth agenesis.
The Msx1 and Pax9 kindred’s have a high but equal probability of missing the third molars and hence the absence of third molars is not a useful indicator of the particular gene (Msx1 or Pax9) that is likely to be affected in a given kindred. Mutations in Msx-1 and Pax-9 genes may cause different types of oligodontia (different sets of teeth are missing in different gene mutations).
For example all individuals with a mutation in Msx-1 lack all second premolars and third molars (and a variable number of other permanent teeth). Typically mutations in the Pax-9 cause agenesis of most permanent molars (and again, a variable number of other permanent teeth). These differences presumably reflect different functions of these genes during development.
Very recently it has been shown that oligodontia and predisposition to cancer are caused by a nonsense mutation in the Axin-2 gene. The Axin-2 gene is located on the chromosome 17. The Axin-2 is a Wnt-signaling regulator. Wnt signaling regulates embryonic pattern formation and morphogenesis of most of the organs. Wnt-signal activity is necessary for normal tooth development.
During tooth development Axin-2 is expressed in the dental mesenchyme, the odontoblasts and the enamel knot. Aberrations of regulation of Wnt signaling may lead to cancer. The nonsense mutation of Axin-2 is not only associated with tooth agenesis but also with colorectal cancer.
Dlx1 and Dlx2 genes play an important role in odontogenic patterning. These genes are important in the development of maxillary molars in mice. But these genes are not required for development of incisors and mandibular molars. The mutation of Dlx1 and Dlx2 in mice leads to failure of development of maxillary molars.
Mode of Inheritance of Tooth Agenesis
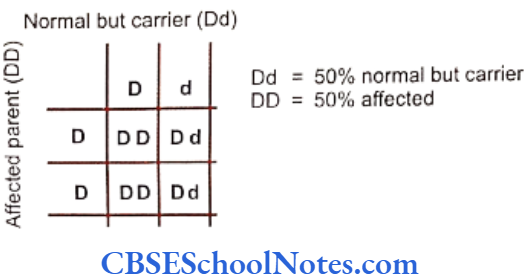
The heritability of developmental missing teeth has been shown in many studies. The responsible genetic factors may be of dominant, recessive or multifactorial (genetic and environment) patterns in terms of inheritance.
Both hypodontia and oligodontia due to mutation in Pax9 and Msx1 genes have autosomal dominant mode of inheritance. However in both the cases the degree and identity of missing teeth may vary between relatives. The variability is probably caused by other genetic and environmental factors and in some cases the etiology is similar to multifactorial traits. Many studies have suggested that most cases of hypodontia have a polygenic inheritance pattern.
In some cases of hypodontia autosomal recessive and X-linked inheritance have also been reported.
Supernumerary Teeth Or Hyperdontia
Hyperdontia is the condition of having supernumerary teeth or teeth which appear in addition to the regular number of teeth.
The most common supernumerary tooth is a mesiodens which is a malformed, peg-like tooth that occurs between the maxillary central incisors. Fourth and fifth molars that form behind the third molars are another kind of supernumerary teeth. Another rare type of supernumerary teeth is a “third set of teeth” that forms underneath and pushes out the second set of teeth, much like the second set that is formed underneath and pushes out the first set of teeth.
Hyperdontia can be syndromic (i.e. associated with Gardner’s syndrome, cleft lip and cleft palate and Hyperdontia can be syndromic (i.e. associated with cleidocranial dysostosis) or nonsyndromic (isolated). Supernumerary teeth in deciduous dentition are less common than seen in permanent dentition.
The etiology of supernumerary teeth is not completely understood. It is thought that the super- numerary tooth is created as a result of a branching of the tooth bud or from fragmentation of the dental lamina. Genetics may also play a defining role in the occurrence of this anomaly (D’souza et al, 2007) as supernumeraries are more commonly found in the relatives of affected children than in the general population (Kawashima et al., 2006).
Many studies have indicated that the anomaly follows a simple mendelian pattern of inheritance (autosomal domi- nanat) (Batra et al, 2005) while some other studies indicate that no such definite pattern of inheritance exist.
Taurodontism
Taurodontism is a condition found in teeth where the body of the tooth and pulp chamber is enlarged at the expense of the root. The changes of taurodontism are usually most striking in the molars.
Taurodontism was a frequent finding in early man and is found today in races such as the Eskimos who use their teeth for cutting hides. The mode of inheritance of the condition is not very specific as it is likely to be polygenic. Few studies have suggested it to be a dominant, others as recessive and some others as an X-linked trait. This condition is also associated with various syndromes, e.g. the trichodento-osseous syndrome, otodental dysplasia and Klinefelter syndrome.
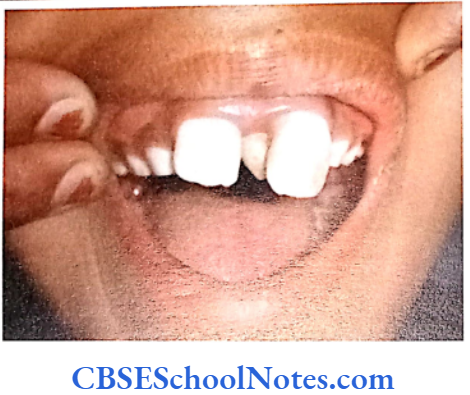
Amelogenesis Imperfecta
When a person’s teeth are covered by thin or malformed enamel, the condition is known as amelogenesis imperfecta (AI). The condition results due to abnormal formation of the enamel. This condition affects both the deciduous and permanent teeth.
Amelogenesis imperfecta is an inheritable (genetic) condition caused by mutation in genes which encode for enamel matrix proteins (Santos et al, 2005). These enamel proteins are needed for formation of normal enamel. As there are many genes (proteins) involved in the formation of enamel, AI presents in many phenotypic forms.
AI is usually classified in four major categories (Crawford et al., 1992) and 14 subtypes.
Characteristics of Various Types of Al
Hypoplastic type of Al
- Enamel formed is hard and well-calcified but its amount is insufficient (incomplete formation of the organic enamel matrix of teeth).
- Phenotypically enamel defect is seen in various forms, viz. as generalized defects (affecting complete enamel thus enamel is thin and translucent) and as localized defects (pits and grooves are seen in the specific areas of the enamel), etc.
- When the enamel is thin the teeth are of small size and as such they may not contact each other mesodistally. As enamel is very thin, teeth are sensitive to thermal stimuli.
- The irregular formation of enamel (absence of enamel in some areas) is due to the absence of ameloblast cells in some areas of the enamel organ.
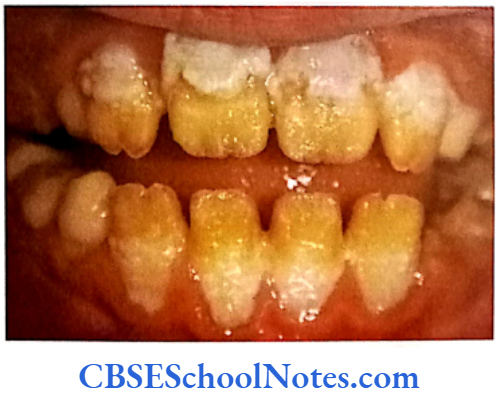
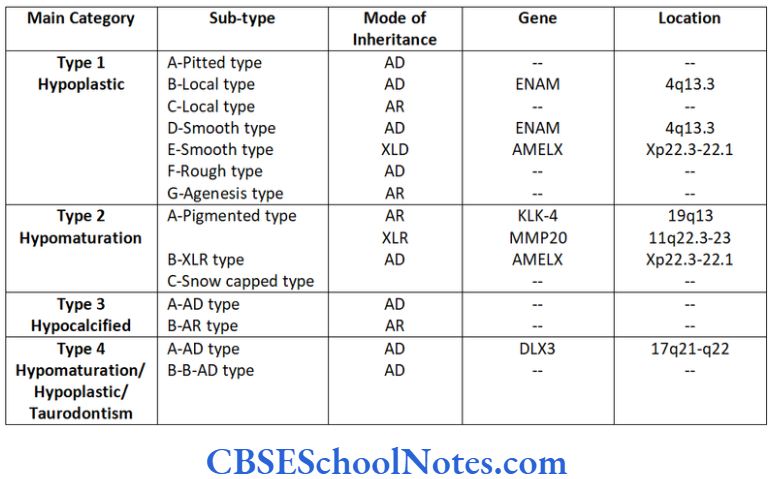
Hypomaturation Type of Al
- Enamel is of normal thickness (not hypoplastic). It is relatively of normal hardness (slightly hypocalcified). Shows reduced radiographic density.
- Enamel is opaque and has a porous surface that becomes stained (white to brownish yellow).
- Teeth are soft and vulnerable to attrition.
Hypocalcified Type of Al
- Enamel matrix is formed but poorly calcified.
- Enamel is of normal thickness, very soft and has a cheesy consistency.
- It is opaque, fragile and chalky in appearance. Teeth tend to become stained.
- It gets chipped away easily during mastication.
- Many teeth may fail to erupt.
Hypomaturation/Hypoplastic/Taurodontism
Type of Al
- The enamel appears mottled.
- Teeth may be pitted on facial surface and are yellowish brown color.
- Molar teeth may show taurodontism.
- Pulp chamber are enlarged in molar teeth.
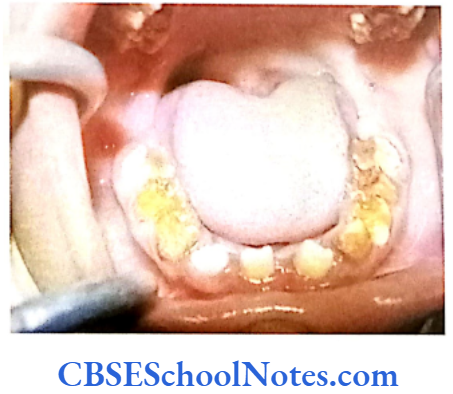
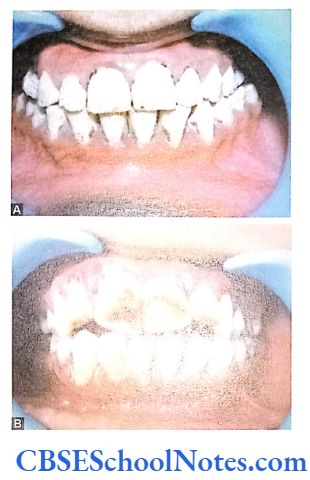
Genetics of Al
How is Al Produced?
The enamel is mostly composed of a mineral (calcium hydroxyapetite) that is formed and regulated by proteins in it. Major proteins that help in the formation AMELX Xp22.3-22.1 Amelogenins Hypoplastic type of Al of enamel are ameloblastin, enamelin, tuftelins and amelogenin. Protein amelogenin is the most abundant protein in enamel (about 90% of all the enamel proteins).
It helps to separate (produces spacing) and support the ribbon-like enamel crystals as they grow. It also regulates the thickness of enamel. Proteins enamelin and ameloblastins are needed to shape and organize the mineral containing crystals in the developing enamel. Ameloblastins are believed to guide the enamel mineralization process by controlling elongation of enamel crystals and to form junctional complexes between enamel crystals.
In the developing enamel ameloblastin consists of 5% and enamelin 2% of total enamel proteins. Tuftelins are located near the dentoenamel junction. They help in the nucleation of enamel crystals. Tuftelins is present in the enamel tufts.
Once the functions of amelogenins and ameloblstins are over in the developing enamel they are cleaved and removed during the maturation stage of the enamel. Two different proteolytic enzymes cleave these proteins namely enamelysin and kallikrein-4.
Kallikrein is responsible for cleavage of the amelogenin protein and enamelysin cleaves amelogenin, ameloblastin and enamelins enamel proteins. Thus after maturation very little protein remains in the enamel (products of cleaved enamelins and tuftelins).
The malformation of these proteins (either due to their absence or altered structure) leads to the formation of abnormal enamel (amelogenesis imperfecta-AI). As genes code all these proteins, mutations in these genes cause AI. Genes like AMELX, ENAM, KLK4, MMP20 and DLX3 code for the major protein components involved in the formation of enamel.
Protein tuftelins is produced by the gene TUFT located on the long arm of chromosome number 1 at position 21st (1q21). The following table gives the details about the genes and proteins coded by them.
Various known genes responsible for AI:
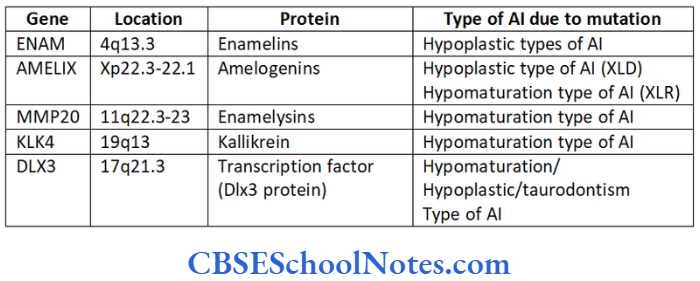
ENAM
This gene codes for the protein enamelin needed for normal development of enamel. The gene is located on the long arm of chromosome number 4 at position 13.3 (4q13.3). Various mutations are identified in the ENAM gene that leads to the production of altered enamelins protein (Hart et al, 2003 and Rajpor et al, 2001).
Sometimes the mutation is significant enough to stop the synthesis of enamelins absolutely. The absence or altered structure of enamelins leads to abnormal development of enamel. It either produces severe defects in the enamel (completely absent enamel or thin enamel) or milder defects (pits, ridges or grooves in the enamel).
The mutations in ENAM gene are inherited as autosomal dominant (AD) or autosomal recessive (AR) in association with hypoplastic type of AI (Kida et al, 2002).
The AMBN gene codes for the protein ameloblastin and is situated close to the ENAM gene. Both genes are located together on the long arm of chromosome 4 at 13th position (4q13). The AMBN gene is also said to be located at the 4q21 position and its mutation leads to AIH2 type of hypoplastic amelogenesis imperfecta.
AMELX
The AMELX gene codes for production of the amelogenin protein. Similar to the ENAM gene many mutations of AMELX gene have been identified. The mutation may lead to a nonproduction or altered production of amelogenin. This interferes with the formation and organization of enamel crystals. Enamel cannot form without adequate amount of amelogenin.
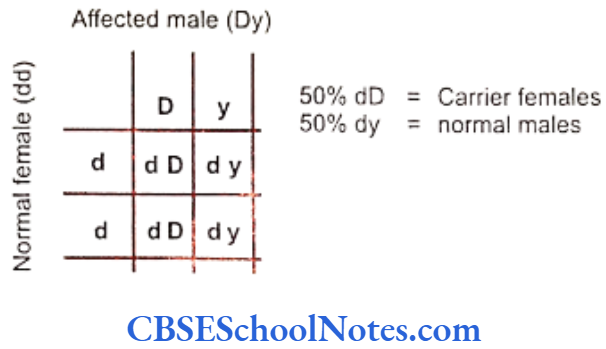
The gene is located on the short arm of X-chromosome between positions 22.31 and 22.1 (Xp22.3 22.1). The mutation is inherited in an X-linked dominant (XLD) and X-linked recessive (XLR) pattern. In case of the XLD disease the males are severely affected owing to complete lack of amelogenin. They develop no enamel to cover their teeth.
However in the case of females (due to the mechanism of Lyonization of X-chromosome) some enamel is always formed in cells where the normal X-chromosome is not inactivated. But this enamel is phenotypically abnormal as it shows structural defects (vertical grooves) in them. The XLD inheritance is observed in hypoplastic type of AI while XLR inheritance is observed in hypomaturation type of AI (Ravassipour et al., 2000 and Hart et al, 2002).
Similar to the AMELX gene on X chromosome, amelogenin producing gene is present on the Y chromosome. It is known as AMELY. This gene is not similar to amelogenin gene on the X-chromosome because it has a different sequence of amino acids. This gene (AMELY) is not important in enamel formation. Only the AMELX gene is critical in enamel development.
The AMELY is situated on the short arm of Y-chromosome at 11th position (Yp11).
MMP20
The MMP20 (Matrix Metallopeptidase 20) gene codes for the protein enamelysin. Enamelysin is needed to cleave other proteins like ameloblastin and amelogenin during the maturation of the enamel. After the cleavage by enamelysin these proteins are easily removed from the enamel.
In case of mutations of MMP20 genes enamelysin is not produced. Thus ameloblastin and amelogenin and other enamel forming proteins are not cleaved and remain present in the developing enamel resulting in soft enamel having an abnormal crystal structure. Hypomature teeth are formed as a consequence.
The gene is located on the long arm of chromosome number 11 at position 22.3 (11q 22.3). The mutation of MMP20 gene is inherited in an autosomal recessive pattern and leads to hypomaturation type (pigmented type) of AI (Li W, et al., 2001).
KLK-4
KLK-4 gene codes for the protein kallikrein, a proteolytic enzyme belonging to the tissue kallikrein family of serine proteases. This enzyme is responsible for the degradation of enamel protein during the maturation stage.
The gene is present on the long arm of chromosome 19 at 13th position (19 q 13). The mutation of this gene leads to hypomaturation (pigmented) type of AI which is inherited as an autosomal recessive trait (Hart et al, 2004).
DLX3
The DLX3 (Distal-less homeobox 3) is a transcription factor gene which codes for the Dlx3 protein. It is a highly penetrant gene whose mutation leads to hypomaturation/hypoplastic / taurodontinism type of AI. The DLX3 gene is located on the long arm of chromosome 17 at position 21.3 (17 q 21.3).
It should be noted that hypocalcified type of AI has not been associated with any specific gene till date.
Dentinogenesis Imperfecta
The term dentinogenesis imperfecta (DGI) is defined as a genetic disease, which leads in the formation of defective dentine. The dentin is poorly formed with an abnormal low mineral content. Here the enamel is normal but the pulp chamber and pulp canal are obliterated.
This condition is also associated with discoloration of teeth (dusky blue to brownish). Teeth usually wear down rapidly leaving short and brown stumps. This problem affects both the primary and permanent teeth.
Incidence: The incidence varies from 1 in 6000 to 1 in 8000 births.
Types of DGI
Shields has described three different types of dentinogenesis imperfecta:
- Shields Type 1 DGI– This type is associated with osteogenesis imperfecta (OI), a condition where bones are congenitally brittle and easily broken. This is an inherited defect of collagen formation which results in weak and brittle bones, bowing of limbs and blue sclera. The milk teeth are more severely affected in this condition. Teeth may show an amber translucent color. Crowns are bulbous and pulp chambers show obliteration in radiographs.
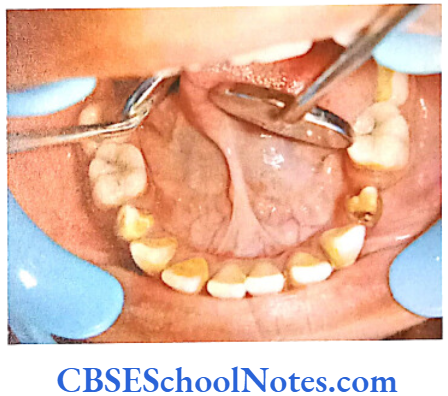
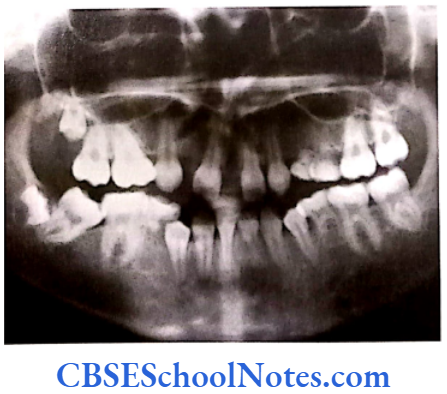
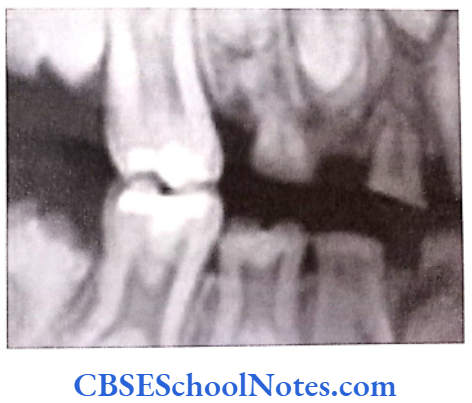
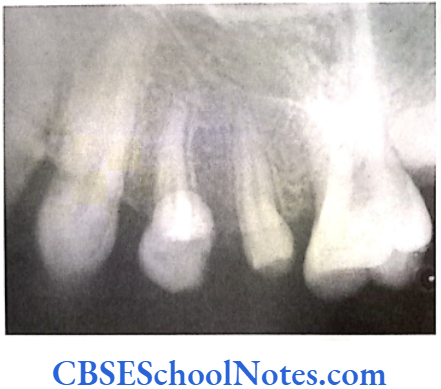
- Shields Type 2 DGI-Dentinogenesis imperfecta is an entity clearly distinct from osteogenesis imperfecta and manifests with opalescent teeth (Fig.10.14) and the teeth are the only structure affected in the body. There is no incidence of increased frequency of bone fractures in this disorder. In this type of DGI only the dentin is affected. Witkop and Rao (1971) preferred the term hereditary opalescent dentin for this condition as an isolated trait and reserving the term dentinogenesis imperfecta for the trait combined with osteogenesis imperfecta. The teeth are blue-gray or amber brown and opalescent. On dental radiographs, the teeth have bulbous crowns, roots that are narrower than normal, and pulp chambers and root canals that are smaller than normal or completely obliterated (Fig.10.15).
This type of DGI is sometimes associated with progressive loss of hearing.
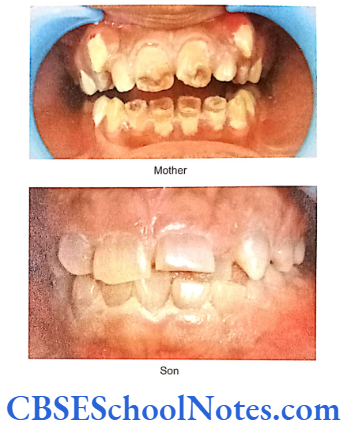
- Shields Type 3 DGI-It is called the brandywine form after the city of Brandywine in Maryland where a large population of patients were affected with this disorder. Similar to the features in type 2 of the disorder, this particular variant affects only the dentine without any involvement of the bones. Type 3 tends to be less severe than type 2 disease. Whether type 3 should be considered a distinct phenotype or a variation of DGI 2 is debatable. Witkop (1975) indicated that the type 2 and type 3 variants might be one and the same because of their clinical similarities. However, unlike the type 2 and type 1 chracteristic, type 3 is associated with shell-like teeth having multiple attrition. Cast metal crowns on posterior teeth and pulp exposures.
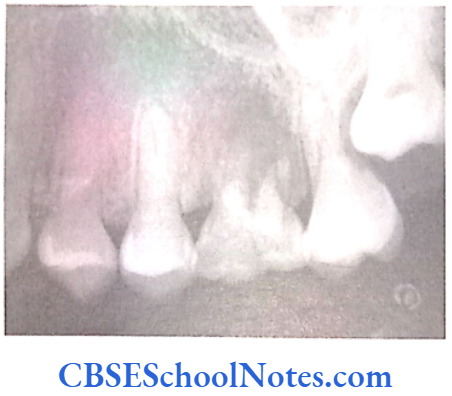
Genetics of DGI
The gene DSPP is responsible for coding dental sialophosphoprotein. Soon after the production of this protein it is cleaved into three smaller proteins namely, the dentin sialoprotein (DSP), dentin glycoprotein and dentin phosphoprotein (DPP) needed for the formation of dentin.
The first two proteins are involved in the normal hardening of collagen. All the three proteins help in the deposition of mineral crystals among collagen fibers (mineralization).
Dentinogenesis imperfecta type 2 is caused due to the mutation in Gene DSPP (Zhang et al, 2001) that codes for dental sialophosphoprotein. Thus the deficiency of dentin sialophosphoprotein causes DGI.
The formation of dentine is defective due to less hardening of collagen, i.e. due to the imperfectly formed matrix. The mineral content in the tissue is less than normal dentine and contains more water (hence soft). This results in discolored teeth which are weak and likely to decay and break.
The DSPP gene is located on the long arm of chromosome number 4 at position 21.3 (4q21.3). About 10 different mutations have been identified in people with DGI. This leads to two forms of DGI, viz. type 2 and type 3 types (Mac Douqall et al, 1999).
The mutation in the DSPP gene is also responsible for causing dentin dysplasia type 2 where there is a substitution in a single amino acid (tyrosine with aspartic acid at protein position 6). As the mutation in DSPP gene gives rise to DGI types 2 and 3 and dentin dysplasia, it indicates that all the three variants are different allelic forms of the same disease.
DSPP gene is also active at low levels in the inner ear and may play a role in normal hearing. Therefore the DGI type 2 is associated with progressive loss of hearing (Xiao et al, 2001).
Dentinogenesis imperfecta is inherited as an autosomal dominant trait and an affected person has one affected parent with DGI.
Treatment
The main aim of treatment is to prevent the loss of enamel. This will further prevent the loss of dentin by jacket crown on anterior teeth may achieve this purpose.
Dentine Dysplasia
Dentin dysplasia (DD) is a genetic disorder of dentin formation with abnormal pulpal morphology. It affects approximately 1 in 100,000 people. Two varieties of dentin dysplasia, type 1 and type 2, have been recognized. Both are inherited in an autosomal dominant manner.
Type 1 DD disorder is also known as radicular dentin dysplasia since the underdeveloped roots and abnormal pulp tissues are predominately located in the roots of the teeth. The deciduous teeth lack pulp chambers or have half-moon shaped pulp chambers in short or abnormally shaped roots.
The condition may affect primary as well as adult teeth. Since the roots are abnormally short, blunt and conical it usually leads to premature loss of teeth. The color of the teeth is generally normal or with slightly amber translucency. The gene or genes responsible for this condition is not known.
Dentin dysplasia type 2 appears virtually identical to dentinogenesis imperfecta type 2 in the primary dentition with yellow-brown to blue-gray discolora- tion of the teeth and pulpal obliteration. However, unlike dentinogenesis imperfecta, the permanent teeth in dentin dysplasia type 2 are normal in color and on radiographs have a thistle-tube pulp chamber configuration with pulp stones.
Genetics of DD type 2
Dentin dysplasia type 2 is due to mutations in the gene DSPP. Due to the similar phenotype of the primary teeth and known similar gene loci (gene DSPP, 4q21.3) for DD type 2 and DGI type 2, it was speculated that DD type 2 could be an allelic variant of mutation in the gene responsible for causing DGI Shields type 2 (Beattie et al, 2006).
Studies have now proved in at least some families that DD type 2 is caused by mutations in the DSPP gene which is associated also with DI type 2.
Management of dentin dysplasia comprises preventive oral health care with meticulous oral hygiene.
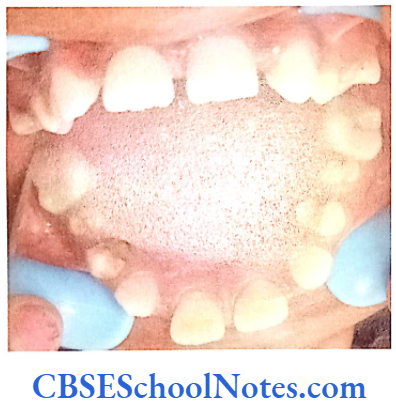
Hypophosphatasia
It is an inherited disease affecting development of bones and teeth. This condition results due to the faulty mineralization (calcium and phosphorus) of bones and teeth. This condition occurs due to the deficiency of the enzyme alkaline phosphatase. This enzyme plays a role is the mineralization of bones and teeth.
This enzyme is encoded by the ALPL gene (alkaline phosphates, liver/kidney/bone). The gene is located on the short arm of chromosome number 1 between the p36.1 to p34 positions.
Persons affected with the severe form of disease may show early loss of primary teeth. Affected child has a short stature with bowed legs or knock-knees, abnormal shape of the skull, enlarged ankle and wrist joints. Affected individuals may also lose their adult teeth permanently.
The mildest form of the disease is called odonto-hypophosphatasia. In this form teeth are the only structures affected and skeletal abnormalities are not observed. People with this condition have abnormal tooth development and premature tooth loss.
Mutation in ALPL gene produces abnormal alkaline phosphatase that results in poor mineralization. In most cases the mutation is mild and actually is a change in a single amino acid. However in cases of severe mutations there may be complete absence of the enzyme.
Mode of Inheritance
The severe form of hypophosphatasia is inherited as an autosomal recessive trait and the milder form by an autosomal dominant pattern of inheritance.