
Genetics Of Craniofacial Disorders And Syndromes
Molecular Regulation Of The Development Of Face
During the beginning of the 4th week of embryonic development the face is represented by stomatodeum situated beneath the developing brain. The stomato- deum is separated from primitive gut by bucco- pharyngeal membrane. This membrane breaks down at the end of 4th week. Surrounding structures of stomatodeum ultimately form the face.
Genetics Of Craniofacial Disorders
These structures are the frontonasal process, the medial and lateral nasal processes, the maxillary and mandibular processes. All these facial processes are covered by the ectoderm beneath which lies the mesoderm (mesenchyme). The mesenchyme of upper face comes from neural crest cells from the forebrain and midbrain areas while that of the mandible come from neural crest cells of midbrain and hindbrain region.
Genetics Of Craniofacial Disorders
The genetic control of the early development of the face is guided as per sequential events given below.
- The frontonasal process is formed by the synthesis of retinoid acid in the ectodermal cells covering the forebrain. Retinoic acid is responsible for the maintenance of the fibroblast growth factor -8 (FGF-8) signals and sonic hedgehog (Shh) signals (in the forebrain and in frontonasal ectoderm) (Carlson, 2004).
- Shh and FGF-8 molecules stimulate neural crest cells to proliferate in the frontonasal process.
- After the 5th week of development the proli- feration of frontonasal process slows down and maxillary, mandibular, medial and lateral nasal processes start growing rapidly. Growth of all these processes results from interactions between the overlying ectoderm and underlying mesoderm. Here again the active signaling molecules in the ectoderm are FGF-8 and Shh. These signals stimulate growth of the mesenchyme.
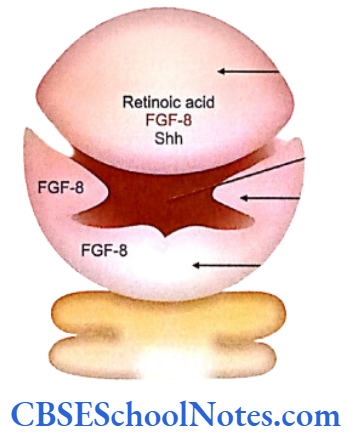
- The growth of the maxillary process is due to establishment of a signaling center in the mandibular arch. FGF- 8 is the molecular signal for maxillary process formation.
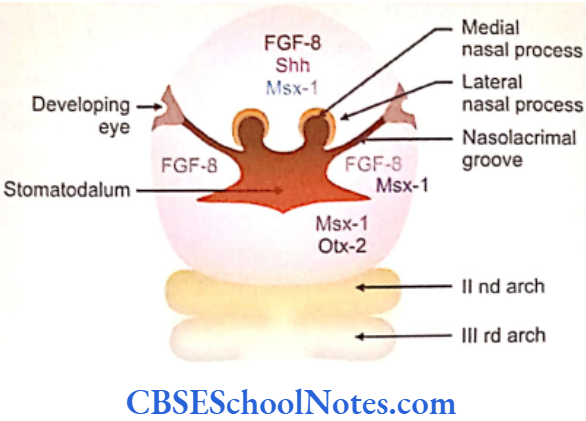
- Next the homeobox containing Msx-1 gene is expressed in the mesenchyme of all the facial processes.
- The transcription factor Otx-2 is expressed in the first arch (maxillary and mandibular processes). This gene characterizes the precursors of the first arch. (It should be noted that the Hox genes are not expressed in the first arch. However, they are expressed in all the other pharyngeal arches).
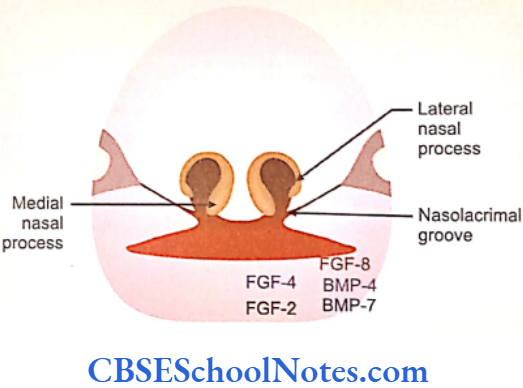
- Further development of the mandibular process is strictly under genetic control. The medial region of the mandibular process responds to FGF-2 and FGF-4 local epithelial signals and stimulates growth of the underlying mesenchyme. These signals are mediated through Msx-1 factors. Growth of lateral region of mandibular process is due to FGF-8 signals. These signals are mediated by bone morphogenetic proteins- Bmp-4 and Bip-7 which are produced in the lateral regions of the mandibular process.
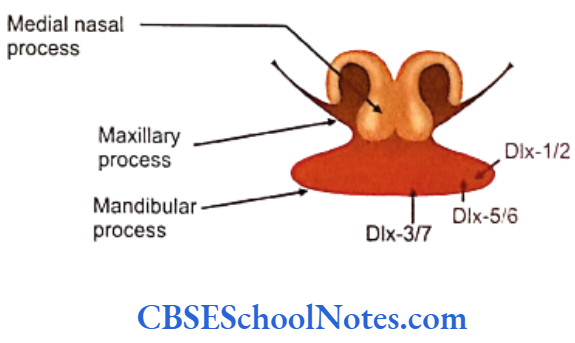
- The development of the mandibular arch in the proximal to distal direction depends upon the expression on Dlx group of transcription factors.
- Dlx-1 and Dlx-2 are expressed most proximally in the mandibular process; Dlx-5 and Dlx-6 are expressed more proximally and Dlx-3 and Dlx-7 are expressed most distally.
- Dlx-1 and Dlx-2 are also expressed in the maxillary process.
Read and Learn More Genetics in Dentistry Notes
During the 7th week the maxillary processes increase in size and move medially. The maxillary process now fuses with the medial nasal process on each side. Hence, the upper lip is formed by the fusion of two maxillary and two medial nasal processes. Stomatodeum is now bound above by the upper lip.
The two mandibular processes grow medially and fuse in the midline to form the lower lip and lower jaw. The fused mandibular processes now form the lower margin of stomatodeum.
To begin with, the lateral nasal process and maxillary process are separated by a deep furrow, the nasolacrimal groove. This groove extends upto the developing eye. The ectoderm of the floor of nasolacrimal groove forms a solid epithelial cord. The cord gets detached from the surface epithelium and gets canalized to form lacrimal sac and nasolacrimal duct at a later stage.
After formation of the basic facial structures by the end of 7th embryonic week, these structures are invaded by mesenchymal cells of the 1st and 2nd pharyngeal arches. The first arch mesenchymal cells form muscles of mastication and are innervated by the trigeminal (5) nerve. The mesenchymal cells of second arch form the muscles of facial expression and are thus innervated by the facial (7) nerve.
Ectodermal Dysplasia
Ectodermal dysplasias (EDs) are heritable conditions characterized by abnormal development of two or more ectodermal structures such as the hair, teeth, nails and sweat glands. Besides these defects related to embryonic ectoderm, an affected person may also show defects of cranial and facial structures, digits and some other parts of the body.
The disease is caused due to defects in the cutaneous and oral embryonic ectoderm and as such it may simultaneously affect many structures that are derived from the ectoderm. Therefore, each person with ectodermal dysplasia may have a different combination of ectodermal defects.
One may have hair and nails affected while another may involve sweat glands and teeth. Each combination is considered a distinct type of ectodermal dysplasia. More than 192 distinct disorders have been defined till date.
Incidence: 1 case per 10,000 births to 1 in 17,000 births.
Pathophysiology
Sweat gland defects-People with ectodermal dysplasia may not sweat or may have decreased sweating due to the lack of sweat gland development. Children with the disease may have defective body mechanisms that control fevers because the skin cannot sweat and control temperature properly. Affected adults are unable to tolerate a warm environment.
Defects of other glands derived from ectoderm– Hypoplasia of the salivary and lacrimal glands may occur. In some patients mucous glands may be absent in the upper respiratory tract and in the bronchi, esophagus, and duodenum. These defects may result in oral dryness, absence of tears and difficulty in swallowing.
Hair defects: Affected individuals tend to have sparse scalp and body hair (hypotrichosis). The hair is often light-colored, brittle and slow-growing.
Abnormal nails-Abnormal nail plate formation may result in brittle, thin, ridged or grossly deformed nails.
Abnormal teeth-Abnormal morphogenesis or absence of multiple teeth may occur.
Additional features-This includes a prominent forehead, thick lips and a flattened bridge of the nose. Sometimes affected person may also show thin, wrinkled and dark-colored skin around the eyes; chronic skin problems such as eczema and a bad- smelling discharge from the nose (ozena).
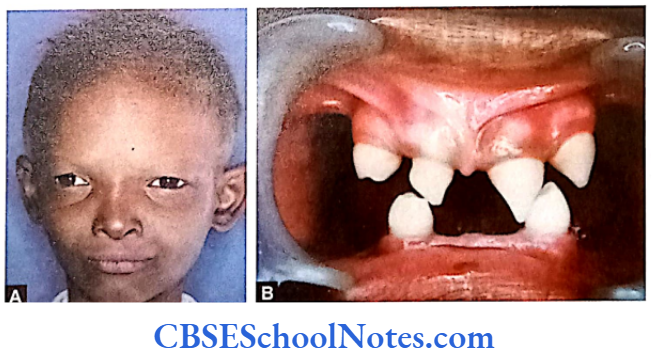
Following are few very common EDs:
Hypohidrotic ED
This is also known as EDA (anhidrotic ED) or Christ- Siemens-Touraine syndrome. It is the most common form of ED. The condition is mostly (in 95% of cases) inherited as an X-linked recessive trait. As a consequence only males are affected and females are carriers. However, due to lyonization and mosaic expression of the abnormal X-chromosome, females may show mild features of EDA.
Less commonly (in 5% cases) the condition is inherited as an autosomal dominant and autosomal recessive disoder.
Hypohidrotic ED Clinical Features
The affected person shows typical facial features represented by a prominent forehead, thick lips, broad nose, sunken cheeks, low set ears and wrinkled and dark eyelids. Teeth are either less in number or completely absent. If present, teeth are conical or pegged-shaped. Patients may have very less, dry and light hair. The skin is dry due to lack of sweat gland formation and these patients may acquire eczematous conditions of the skin. Tears are often absent.
Genetics of Hypohidrotic ED
The EDA, EDAR, and EDARADD genes provide instructions for manufacturing proteins that work together during embryonic development. These proteins form the parts of a signaling pathway that is critical for interaction between the two cell layers, the ectoderm and the mesoderm.
In the early embryo these cell layers form the basis for the genesis of many of the body’s organs and tissues. Ectoderm- mesoderm interactions are essential for the formation of several structures that arise from the ectoderm including the skin, hair, nails, teeth, and sweat glands.
The EDA gene provides instructions for making a protein called ectodysplasin A (Bayes, et al 1998 and Monreal, et al 1998). This protein is part of a signaling pathway that plays an important role in the development of ectodermal appendages (hair, teeth and sweat glands) before birth. The ectodysplasin-A has an important role to play in ectodermal- mesodermal interactions during embryonic development.
Defects in the molecular structure of this protein inhibit the action of enzymes necessary for normal development of ectoderm (Chen, et al 2001). More than 60 mutations have been identified in the EDA gene. The gene is located on the long (q) arm of the X-chromosome between positions 12 and 13.1 (Xq12-q13.1). The hypohydrotic ED is inherited as an X-linked recessive trait.
The EDAR gene provides instructions for making a receptor protein called the ectodysplasin A receptor. This protein is a part of the signaling pathway that plays an important role in development before birth. The ectodysplasin A receptor interacts with a protein called ectodysplasin A (produced from the EDA gene).
Ectodysplasin A attaches to this receptor on the cell surface like a key in its lock. When these two proteins are connected they trigger a series of chemical signals that affect cell activities such as division, growth, and maturation. Gene EDR is located on long arm of chromosome number 2 between 11 and 13 positions. Mutations of this gene cause hypohidrotic ectodermal dysplasia which is inherited as autosomal recessive trait (Shimomura, et al 2004).
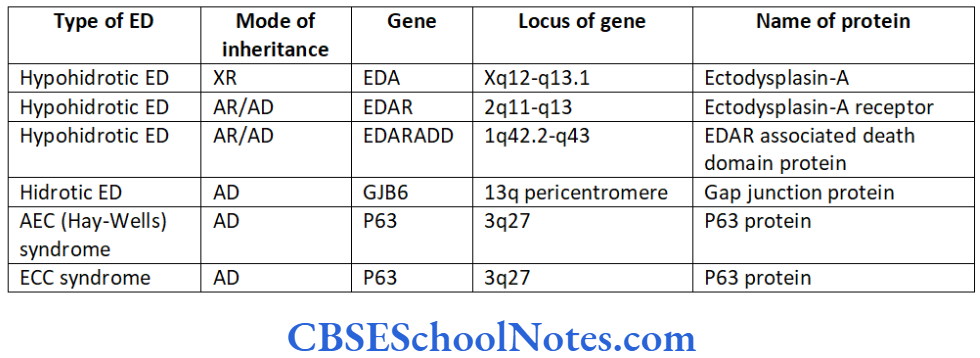
The EDARADD gene provides instructions for making a protein called the EDAR-associated death domain protein (Headon, et al 2001). This protein is part of a signaling pathway active in developmental events before birth. The EDARADD protein interacts with another protein, called the ectodysplasin A receptor which is produced from the EDAR gene. This interaction occurs at a region called the death domain.
This domain configuration is present in both the proteins. The EDARADD protein acts as an adapter, which means it assists the ectodysplasin A receptor in triggering chemical signals within cells. These signals affect cell activities such as division, growth and maturation. Before birth this signaling pathway controls the formation of ectodermal structures such as hair follicles, sweat glands, and teeth.
The EDARADD gene is located on the long arm of chromosome number 1 at 43th position (1q43). Mutation of this gene causes hypohidrotic ED, which is inherited as autosomal recessive inheritance.
Mutations in the EDA, EDAR, or EDARADD gene prevent normal interactions between the ectoderm and the mesoderm and thus impair the normal develop ment of hair, sweat glands and teeth. The improper formation of these ectodermal structures leads to the characteristic features of hypohidrotic ectodermal dysplasia.
Some mutations in the EDA gene represent alterations or substitutions in single DNA building blocks (base pair parameters) whereas other mutations insert or delete genetic material in the gene. These changes lead to the production of a nonfunctional version of the ectodysplasin A protein.
This abnormal protein cannot trigger chemical signals needed for normal interactions between the ectoderm and the mesoderm. Without these signals hair follicles, teeth, sweat glands and other ectodermal structures do not form properly leading to the characteristic features of hypohidrotic ectodermal dysplasia.
Hidrotic ED
This condition is also known as the Clouston syndrome. It is inherited as an autosomal dominant disease. The gap junction proteins help in the communication and interaction between cells. The hidrotic ED mutation is present in the GJB6 gene located in the long arm of chromosome number 13 in its pericentriolar region (13q). This gene encodes the gap junction protein connexin 30 (Cx30) (Jerome, et al 2000 and Guilherme, et al 2004).
Hidrotic ED Clinical features
Scalp hair is very sparse, fine and brittle. Alopecia is common. Eyebrows are thinned or absent. Nail dystrophy is commonly seen. Persistent paronychial infections are frequent. Fingers and toes are abnormal, i.e. they are either more in number or fused with each other. Bulbous fingertips may be present. Patients may have normal face, normal teeth and normal sweating.
AEC (Hay-Wells) Syndrome
Its full name is ankyloblepharon-ectodermal dysplasia- clefting. It is inherited as an autosomal dominant trait of variable expressivity. It is due to a mutation in the p63 gene, which is located on the long arm of chromosome number 3 at position 27. The syndrome is caused by heterogenous missense mutation in p63 (McGrath, et al 2001).
AEC (Hay-Wells) Syndrome Clinical Features
Patients exhibit characteristic facial features like ankyloblepharon (congenital adhesion of the upper and lower eyelids by fibrous bands), sunken maxilla, broad nasal bridge and cleft palate. Absence or sparse hair in the scalp, absence or malformation of nails and pegged teeth are presented in the disease. Mild hypohydrosis is also common.
EEC Syndrome
EEC is an acronym of ectrodactyly-ectodermal dysplasia-cleft lip/cleft palate syndrome. It is inherited as an autosomal dominant trait of low penetrance and variable expressivity. The disease is due to a mutation in the gene p63 located on long arm of the chromosome number 3 at position 27 (Bokhoven, et al 2001).
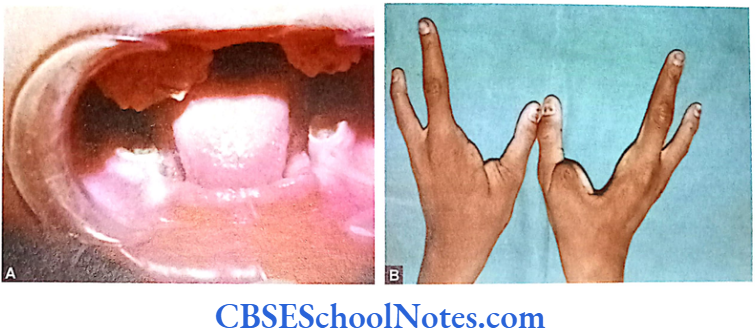
EEC Syndrome Clinical Features
Affected persons may show severely malformed hands and feet (lobster claw deformity). Some may show hypoplastic metacarpals and metatarsals. Cleft lip and palate are very common.
Other features include dry and coarse hair; abnormal nails, mild hypohydrosis, enamel hypoplasia and microdontia.
Associated defects include blepharophimosis, deafness and abnormalities of the genitourinary tract.
EEC Syndrome Treatment
- Wigs can be used or special hair care taken for hair loss.
- Patient should always remain in a cool environment to prevent dryness of skin. Skin care products should be used to prevent dryness of the skin and for prevention from exposure to heat.
- Dental evaluation should be conducted every 6 to 12 months. Dental treatment like simple restoration of dentures or dental implants may become necessary in many patients.
Holoprosencephaly
The face and the forebrain (prosencephalon) of an embryo normally begin to develop in the 5th and 6th weeks of pregnancy. The development of face is related to the development of the forebrain. If the forebrain fails to segment into normal right and left hemispheres, it may result in deformities of the face. The abnormal development of brain and face is known as holoprosencephaly (HPE).
Following three types of the HPES are described in the literature:
Alobar-This type of HPE is features complete failure of separation of right and left cerebral hemispheres. There is single lobe and a single ventricle. This is a very severe form of HPE and the affected usually die during intrauterine life or in their early infancy. This variety is associated with severe craniofacial abnormalities.
Semilobar-This type of HPE is characterized by incomplete separation of cerebral hemispheres. This is associated with milder facial abnormalities.
Lobar-It is marked by substantial but still incomplete separation of the hemispheres. Separation is seen posteriorly but part of frontal lobe may remain fused together. Sometimes patients may have a nearly normal brain.
The diagnosis of the various types of HPE can be easily made with the help of a CAT scan or an MRI.
Holoprosencephaly Signs and Symptoms
The most severe forms of holoprosencephaly produce seizures and mental retardation. Midline structures of brain like the corpus callosum and septum pellucidum are not developed. The two thalamic lobes may be fused into one. Olfactory tracts and bulbs may remain absent in some cases. The circle of Willis is not well developed usually.
Typical facial defects involve the eyes, nose and upper lip. In some cases the nose may be entirely missing. However, there is no strict association between the types of HPE and the degree of severity of the facial defects in each of the types. Yet some associations are commonly seen:
- Facial defects associated with alobar type of HEP-The most severe of the facial defects is cyclopia; an abnormality characterized by the development of a single eye located in the area normally occupied by the root of the nose and a missing nose or a nose in the form of a proboscis (a tubular appendage) located above the eye. Premaxillary agenesis with median cleft lip, ocular hypotelorism, flat nose and sometimes bilateral cleft lip may be present with cyclopia.
- Facial defects associated with semilobar type of HPE-Bilateral cleft lip with the median process representing the philtrum-premaxilla anlage. There may be midline cleft lip and/or palate. Nose may show a flat nasal tip, absence of nasal septum and/or a flat nasal bridge. Ocular hypotelorism is very common. Microcephaly is also seen in many affected persons. Sometimes a person affected with semilobar type of HPE may have relatively normal facial appearance.
- Facial defects associated with lobar type of HPE-The least severe presentation in the spectrum of facial anomalies is the median cleft lip which is also called premaxillary agenesis. Bilateral cleft lip with a median process, ocular hypotelorism and a flat nose may be the only facial abnormalities. Person may also present with relatively normal facial appearance.
Microforms of HPE that can be observed in relatives of probands with HPE include the following:
- Microcephaly/Single central maxillary incisor/ Ocular hypotelorism/Anosmia/hyposmia (resulting from absence of olfactory tracts and bulbs)/Iris coloboma/Absent superior labial frenulum/Midface hypoplasia.
Causes of HPE
Etiology of HPE is heterogeneous. It is due to the fact that both environmental and genetic factors have been identified as causative agents.
HPE Environmental Causes
- Maternal diabetic mellitus during pregnancy • Alcohol
- Retinoic acid
- Hypocholesterol.
HPE Genetic Causes
- Chromosomal Abnormalities
- Trisomy 13 (Patau’s syndrome) and Trisomy 18 (Edward’s syndrome)
- Deletion and duplication of various chromosomes.
- Single Gene Abnormalities
The mutation in a single gene causes two types of HPE, viz. Syndromic HPE (associated with various syndromes) and nonsyndromic HPE.
Nonsyndromic HPE
The nonsyndromic gene mutations responsible for HPE are usually inherited as autosomal dominant traits. The following table indicates genetics of autosomal dominant nonsyndromic HPES
SHH-The human sonic hedgehog gene (SHH) encodes a secreted protein (sonic hedgehog protein). This protein is involved in establishing cell fates at several points during development. It is expressed in the Hensen’s node and floor plate of the neural tube. Thus it is one of the primary inducers of the ventral neural tube.
Various types of mutations of the SHH gene are known (frame shift, heterozygous deletion, miss-sense mutations, etc). Mutation of sonic hedgehog gene leads to faulty production of sonic hedgehog protein that ultimately results in abnormal development of the forebrain and face (Roessler, et al 1996).
ZIC2-This gene encodes zinc finger protein 2. It plays a major role in mediating the response to sonic hedgehog protein signaling. Mutation of ZIC2 causes holoprosencephaly (Brown, et al 1998).
SIX3-This gene encodes the homeobox protein SIX3. It participates in midline forebrain structuring and in formation of eyes. It is present in the anterior region of the neural plate and midline ventral forebrain. Its mutation interferes with the development of brain and eye (Wallis, et al 1999).
TGIF-It modulates the TGIF beta pathways. Thus TGIF links the NODAL signaling pathway leading to the bifurcation of the human forebrain and the establishment of ventral midline structures (Gripp, et al 2000).
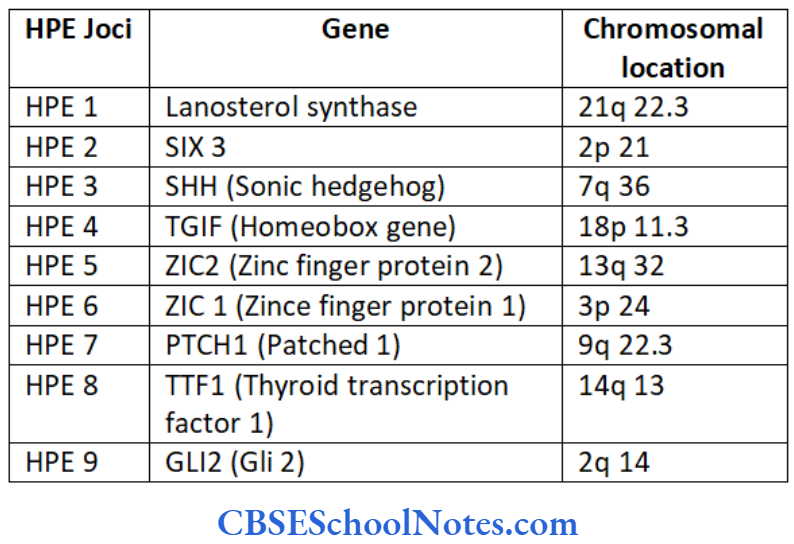
PTCH 1 – This gene encodes for the patched 1 receptor protein. This protein is the receptor for SHH.
Treatment of HPE
The treatment of persons suffering from HPE is basically the management of its symptoms.
- Hormonal replacement in case of pituitary dysfunction.
- Antiepileptic drugs for seizures.
- Surgical repair of abnormalities associated with face such as cleft lip/palate.
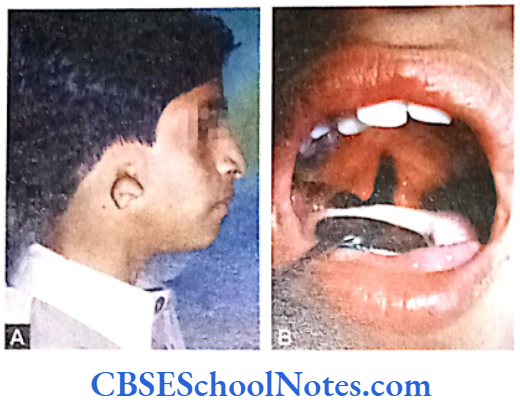
Mandibulofacial Dysostosis (Treacher Collins-Franceschetti Syndrome)
The syndrome is characterized by disorders of craniofacial developmental events. The disease is hereditary in nature.
Mandibulofacial Dysostosis Incidence: From 1 in 40,000 to 1 in 70,000 live births.
Mandibulofacial Dysostosis Clinical Features
Some of the important clinical features are as under:
- Down slanting palpebral fissure, coloboma of lower eyelids.
- Hypoplasia of malar bones and occasional absence of palatine bones.
- Hypoplasia or sometimes agenesis of mandible. Microstomia.
- Cleft palate and malocclusion of teeth.
- Malformation of the external ear. Sometimes middle and internal ears (auditory ossicles, cochlear and vestibular apparatus) are also affected. This leads to deafness.
- Occasional heart defects.
- Patients are of normal intelligence and have normal reproductive life.
Mandibulofacial Dysostosis Genetics
Inheritance: The inheritance shows an autosomal dominant (AD) pattern with complete penetrance and variable expressivities.
The syndrome is due to the mutation in the gene called the Treacher Collins-Franceschetti syndrome 1 (TCOF1) gene. It is located on the long arm of chromosome number 5 (5q31.3-q33.3). Approximately about 150 mutations have been identified in the TCOF 1 gene.
The TCOF1 gene codes for a protein called treacle which is required for normal craniofacial development. Mutation of gene leads to the absence or reduced production of treacle in the cells (Edwards, et al 1997). Researchers believe that a loss of this protein triggers certain apoptotic signals in cells important for the development of facial bones that self-destruct themselves (undergo apoptosis).
This abnormal cell death leads to Treacher Collins syndrome. As per other views towards the syndrome, it occurs due to the failure of migration of neural crest cells in the 1st and 2nd branchial arches. This leads to dysplasia of musculoskeletal derivatives of these arches.
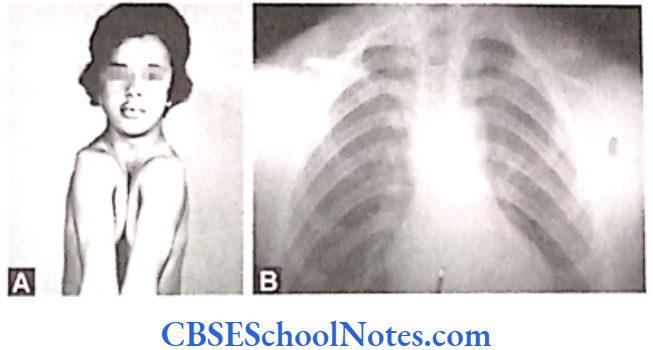
Cleidocranial Dysplasia
Cleidocranial dysplasia (CCD) is a disorder exhibiting defective endochondral and intramembranous bone formation.
Cleidocranial Dysplasia Incidence: 1 per million individuals worldwide.
Cleidocranial Dysplasia Clinical Features
Following abnormalities of bone formation are Inheritance: The CCD is an autosomal dominant observed.
Skull
Closure of the fontanels is delayed with the presence of many sutural bones are in the cranial sutures. Though head is brachycephalic in shape, bossing of frontal, parietal and occipital regions gives the skull a large globular shape.
Shoulder Girdle
Clavicles are either completely or partially absent. The patient can approximate the two shoulders in front of the chest (Fig. 11.8). Sometimes anomalies in other bones of the body may also be seen, e.g. in the pelvis, vertebral column, etc. with shortening of metatarsals, metacarpals and long bones.
Face
Maxilla, lacrimal and zygomatic bones are under- developed. Cleft palate is occasionally present. Paranasal air sinuses are underdeveloped. Sometimes the patients may have enlarged mandible as compared to the normal.
CCD patients may show a delayed eruption of permanent teeth. They may have supernumerary or impacted teeth. Patients are of normal intelligence but slightly short in height as compared to other normal members in the family.
Genetics
Inheritance: The CCD is an autosomal dominant (AD) disorder.
Narahara et al (1995) observed CCD in association with a translocation defect involving chromosomes 6 and 18. It is now believed that CCD is due to a mutation of the gene called core binding factor alpha-1 (CBFA1). This gene belongs to the RUNT transcription factor family and hence the other name of this gene is also runt related transcription factor 2 (RUNX2).
The gene CBFA1 is located on the short arm of chromosome number 6 (6p21) (Mundlos, et al 1995 and Mundlos, et al 1997). CBFA1 gene controls differentiation of precursor cells into osteoblasts and is thus essential for membranous as well as endochondral bone formation.
Researchers believe that the CBFA1 protein acts as a “master switch” regulating a number of other genes involved in the development of cells that build bones (osteoblasts). Thus CBFA1 gene plays an important role in osteoblastic differentiation.
Some mutations result in change of just a single building block (amino acid) of the CBFA1 protein (nucleotide alteration). Other mutations introduce a premature stop signal that result in an abnormally short protein. Occasionally the entire gene may be missing due to an anomaly.
This leads to the shortage of the functional protein (haploinsufficiency of CBFA1) that interferes with normal bone and cartilage development resulting in the signs and symptoms of cleidocranial dysplasia. Thus it may be concluded that CCD is caused due to mutations in the CBFA1 gene and that a heterozygous loss of function (autosomal dominance) is sufficient to cause the disease.
Apert Syndrome (ACROCEPHALOSYNDACTYLY)
It is a congenital genetic defect, which affects the first branchial arch (the precursor of maxilla and mandible). This syndrome is characterized by:
- Craniosynostosis (premature fused cranial suture).
- Craniofacial anomalies like midface hypoplasia
- Syndactyly (fused fingers and toes).
Apert Syndrome Incidence: 1 per 2, 00,000 live births.
Apert Syndrome Clinical Features
- Craniosynostosis involves the coronal suture resulting in a prominent forehead, flat occiput and brachycephaly.
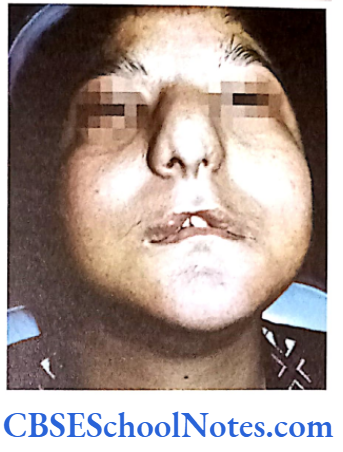
- Down slanting palpebral fissure.
- Low set ears with occasional loss of hearing. • Hypertelorism and exophthalmos.
- Depressed nasal bridge and a short wide nose.
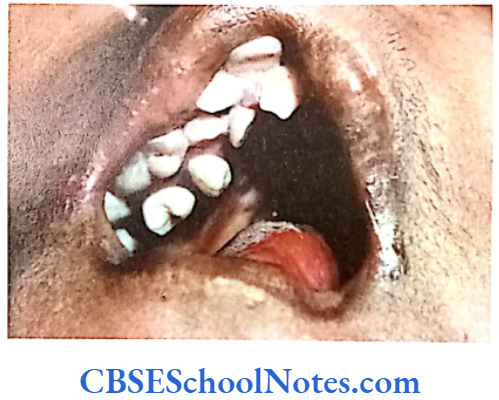
- Maxilla is not well-developed. High arched palate.
- V-shaped maxillary dental arch, malocclusion, supernumerary teeth (Fig.11.10), cleft palate or bifid uvula.
- The digits of hands and feet are either webbed or fused (syndactyly). The severity of the fusion varies with a minimum 3 of digits on each hand the premature fusion of the coronal, sagittal and and foot fused.
- Cardiovascular and GIT anomalies are common.
- Patients may be of normal intelligence but usually retarded.
Apert Syndrome Genetics
The inheritance of the syndrome is autosomal dominant (AD) in nature or the disorder arises fresh due to new mutations in an individual.
Apert syndrome results from mutations in the fibroblast growth factor receptor 2 (FGFR2) genes. This gene is located on the long arm of chromosome number 10 (10q 26). 98% of mutations of FGFR2 genes are substitution mutations such as Ser 252 Trp (between Serine and Tryptophan), Pro 253 Arg (Proline and Arginine) (Wilkie, et al 1995).
Such fibroblast growth factor receptor 2 mutations cause an increase in the number of precursor cells of osteogenic pathways. This again leads to increased subperiosteal bone formation and premature ossification of the calvaria ending in early closure of the cranial sutures.
Syndactyly of Apert syndrome may be due to keratinocyte growth factor receptor (KGFR) mediated effect. The optic disk pallor is more severe in Pro 253 Arg mutation while cleft palate and visual impairment is seen more in the Ser 252 Trp substitution.
Sometimes Apert syndrome may also be caused by deletion/translocation of the short arm of chromosome 2 to long arm of chromosome 11 or 12.
Some other syndromes like Pfeiffer syndrome and Crouzon syndrome (craniofacial dysostosis) also show features of craniosynostosis. These are allelic disorders with overlapping features between the related disorders (Wilkie, et al 1995).
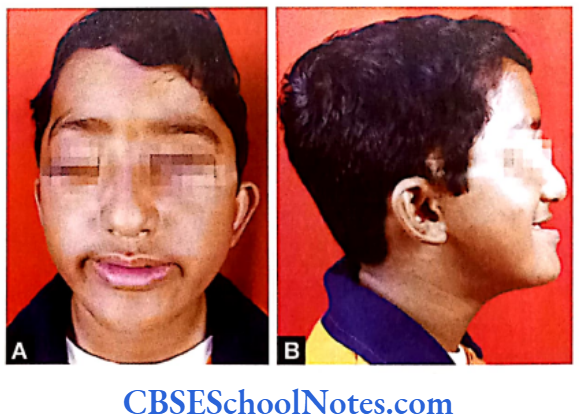
Crouzon Syndrome (Craniofacial Dysostosis)
Crouzon syndrome or craniofacial dysostosis is similar to the Apert syndrome except that it is not associated with syndactyly. Crouzon syndrome is a genetic disorder also known as branchial arch syndrome. Specifically this syndrome affects the first branchial (or pharyngeal) arch; structures that are precursors of the maxilla and mandible.
The main feature of this syndrome is its craniosynostosis or occasionally the lambdoid sutures. The pathological fusion begins in the first year of life and is completed by the second or third year. The order and rate of fusion in the sutures determine the degree of deformity and disability.
Crouzon Syndrome Incidence: 1 in 25000 persons.
Crouzon Syndrome Clinical Features
- Signs of the disease originate from the early closure of cranial sutures.
- Coronal and sagittal sutures are obliterated.
- Short and broad head (brachycephaly).
- Bulging eyes (exophthalmos) due to shallow eye sockets after early fusion of surrounding bones of the orbit.
- Hypertelorism (greater than normal distance between the eyes), divergent squint.
- Progressive optic nerve atrophy results from raised intracranial tension which leads to subsequent visual impairment.
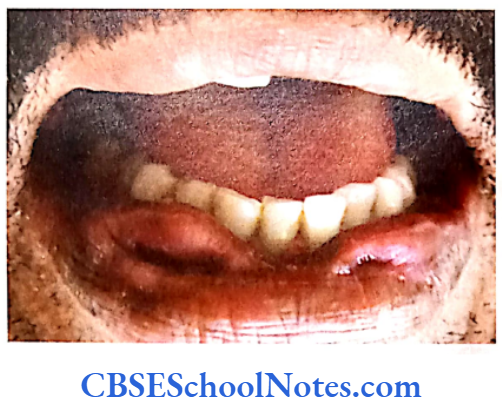
- Under-development of maxilla (insufficient growth of the midface results in protrution of the chin despite normal growth of the mandible). Due to maxillary hypoplasia Crouzon patients generally have a considerable and permanent underbite and subsequently cannot chew using their incisors (they do not use their incisors to take a bite from a sandwitch). For this reason the Crouzon patients eat in an unusual way.
- They may show a narrow/high-arched palate, posterior bilateral crossbite, hypodontia (missing teeth), malocclusion and an increased spacing between the teeth.
- Most Crouzon patients also have noticeably shorter humerus and femur bones in proportion to the rest of their bodies when compared to members of the general population.
- Deafness.
- Mental retardation is a frequent feature.
Crouzon Syndrome Genetics
Crouzon Syndrome Inheritance – Autosomal dominant (AD). Sometimes the disorder arises due to new mutations.
The syndrome is due to a mutation in the fibroblast growth factor receptor genes (FGFR-2) which is mapped to the chromosome locus 10q25-10q26. This gene provides instructions for furnishing a protein called the fibroblast growth factor receptor 2.
Among its multiple functions this protein signals transfor- mation of immature cells into bone cells during embryonic development. Mutations in the FGFR2 gene probably over-stimulate signaling by the FGFR2 protein and result in the bones of the skull fusing prematurely (Rutland, et al 1995).
Crouzon syndrome with acanthosis nigricans is always due to an Ala 391Glu mutation within the transmembrane region of the FGFR3 gene. Acanthosis nigricans is a brown to black, poorly defined, velvety hyperpigmentation of the skin.
Crouzon syndrome exhibits locus heterogeneity with causal mutations in FGFR2 and FGFR3 in different affected individuals.
Crouzon Syndrome Treatment
Surgery is typically used to stop the closure of sutures and prevent damage to the growing brain by compression exerted by the non-expansible skull. Blindness and mental retardation are typical outcomes in neglected cases. Once treated for the cranial vault symptoms, Crouzon patients generally live a normal lifespan.
Pfeiffer Syndrome
Pfeiffer syndrome is a genetic disorder characterized by premature fusion of certain skull bones (cranio- synostosis). This early fusion prevents the skull from growing normally and affects the shape of the head and face. Pfeiffer syndrome also affects bones in the hands and feet.
Pfeiffer Syndrome Incidence: 1 in 100,000 individuals.
Pfeiffer Syndrome Clinical Features
- Pfeiffer syndrome result from the premature fusion of the skull bones.
- High forehead.
- Wide set eyes.
- Underdeveloped upper jaw (maxillae).
- Dental problems due to crowded teeth and often a high palate.
- A beaked nose.
- Poor vision.
- Hearing loss in about 50% of children.
- The thumbs and great toes are wide (broad) and bend away from the other digits.
- Unusually short fingers and toes (brachydactyly) are also common and there may be some webbing or fusion between the digits (syndactyly).
Pfeiffer syndrome is divided into three subtypes as type 1, type 2 and type 3. Type 1 or “classic” Pfeiffer syndrome includes individuals with mild manifes- tations including brachycephaly, midface hypoplasia and finger and toe abnormalities. Most individuals with type 1 have normal intelligence and a normal lifespan.
Types 2 and 3 are more severe forms of Pfeiffer syndrome often involving problems with the nervous system. Type 2 consists of a cloverleaf-shaped skull, extreme proptosis, finger and toe abnormalities, elbow ankylosis or synostosis, developmental delay and neurological complications.
Type 3 is similar to type 2 but without a cloverleaf skull. Clinical overlap between the three types may occur.
Pfeiffer Syndrome Genetics
Pfeiffer Syndrome Inheritance: It is inherited as an Autosomal Dominant (AD) entity.
Mutations in the FGFR1 and FGFR2 genes cause Pfeiffer syndrome.
FGFR1 gene is located on the short arm of chromosome number 8 (8p11.2-p11.1) and FGFR2 is located on the long arm of chromosome number 10 (10q26) (Lajeunie, et al 1995).
The FGFR1 and FGFR2 genes play an important role in signaling the cell to respond to its environment perhaps by division or cell maturation. A mutation in either of the genes causes a prolonged signaling promoting an early maturation of bone cells in a developing embryo and premature fusion of bones in the skull, hands and feet.
Type 1 Pfeiffer syndrome is caused by mutations in either the FGFR1 or FGFR2 genes. Types 2 and 3 are affected by mutations in the FGFR2 gene. Mutations in the FGFR1 usually present a milder phenotype of the disease.
Pfeiffer Syndrome Treatment
Treatment includes multiple-staged surgery of craniosynostosis. Midfacial surgery is performed to reduce the exophthalmos and the midfacial hypoplasia.
Differential Diagnosis
The main differential diagnoses include syndromes that are characterized by craniosynostosis. Mutations in the same FGFR (FGFR1, FGFR2 or FGFR3) can result in different variants of craniosynostosis syndromes thereby implicating a common pathological mecha- nism with a common FGFR gain of function mecha- nism resulting in Pfeiffer, Apert, Muenke, and Beare- Stevenson syndromes.
- Pfeiffer and Apert syndromes are noteworthy for some similarities between them but the two disorders essentially are genetically distinct entities.
- Crouzon syndrome has similar phenotype as the Pfeiffer syndrome but lack the hand and foot anomalies.
- Phenotypic overlap occurs with Muenke syndrome which is caused by a specific FGFR3 mutation.
Cherubism
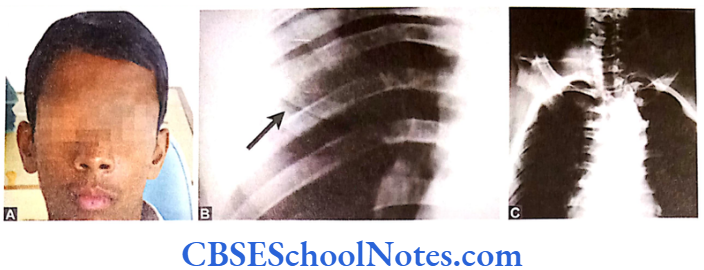
Cherubism is a rare genetic disorder characterized by abnormal bone tissue in the lower part of the face.
Cherubism Incidence: The condition is very rare.
Cherubism Clinical Features
The disease begins in early childhood. Swelling of the lower face starts around the third or fourth year of life and progresses until the late teens. The lower jaw (mandible) and upper jaw (maxilla) become enlarged as the bone is replaced by painless cyst like growths. These growths give the cheeks a swollen and rounded appearance. X-ray reveals multilocular cystic changes in the mandible and maxilla.
The abnormal growths are gradually replaced with normal bone in early adulthood. As a result many affected adults have a normal facial appearance. The deciduous dentition may be shed spontaneously and prematurely. The permanent teeth are also defective in their number that may be more or less than the normal. Most people with cherubism have almost no signs and symptoms affecting other parts of the body.
Cherubism Genetics
Cherubism Inheritance: The mode of inheritance in cherubism is autosomal dominant (AD). Usually the affect person has only a single affected parent or some time the disease is due to new (de novo) mutation.
Cherubism is due to mutation in the SH3BP2 (SH3-domain binding protein 2) gene located on the short arm of chromosome number 4 at the 16.3 position (4p16.3). About 11 different mutations are known in the SH3BP2 gene which give rise to cherubism (Ueki, et al 2001).
Mutations in the SH3BP2 gene lead to the production of an overactive protein. The overactive protein likely causes inflammation in the jaw bones and triggers the production of osteoclasts, which are bone eating cells that breakdown bone tissue during bone remodeling.
An excess of these osteoclasts contributes to the destruction of bone in the upper and lower jaws. A combination of bone loss and inflammation likely underlies the cyst-like growths so characteristic of cherubism.
Cherubism Treatment
Treatment of cherubism is not well-established. It is usually a self limiting disease. Surgical treatment should be designed on individual basis depending on the functional and esthetic need of the patient.
Van Der Woude Syndrome
Van Der Woude Syndrome syndrome is sometimes also known as lip-pit IRF6 protein is active in cells that give rise to tissues syndrome.
Van Der Woude Syndrome Clinical Features
- People with this disorder are born with a cleft lip, a cleft palate or both.
- Affected individuals usually have depressions (pits) near the center of the lower lip which may appear moist due to the presence of salivary and mucous glands in the pits.
- Some individuals may show mucous cysts of lower lip.
- It is a common form of syndromic cleft lip and palate and accounts for 2% of all cleft lip and palate cases.
- Affected individuals may have hypodontia and usually possess normal intelligence. Incidence: The incidence of this syndrome is 1 in 35,000 to 1,00,000 individuals worldwide.
Van Der Woude Syndrome Genetics
Inheritance: Van Der Woude syndrome (VDWS) is inherited as an autosomal dominant (AD) trait or sometimes of sporadic inheritance. The syndrome has 80% penetrance and variable expression.
It is caused by mutations of the IRF6 (interferon regulatory factor 6) gene located on the long arm of chromosome 1 (1q32-q41). About 60 different mutations of IRF6 gene are known currently. The IRF6 protein is active in cells that give rise to tissues in the head and face. It is also involved in the development of other parts of the body including the skin and genitals.
Mutations in the IRF6 gene that cause van der Woude syndrome prevent one copy of the gene in each cell from producing the functional protein. A shortage of the IRF6 protein affects the development and maturation of tissues in the skull and face. These abnormalities underlie the signs and symptoms of van der Woude syndrome that include mouth) and pits or mounds in the lower lip.
The cleft lip, cleft palate (an opening in the roof of the action of certain modifier genes on IRF6 function. marked phenotypic variation may be caused by the Intriguing linkage studies have suggested that a 17p11.2-p11.1 may influence the degree of phenotypic second modifying gene mapped to chromosome expression of a gene defect at this locus.
Most reported cases of VWS have been linked to chromosome 1q32-q41 (VWS1) but a second VWS locus (VWS2) has been mapped to 1p34 (Koillinen, et al 2001). Direct sequence analyses of genes in this region gene encoding interferon regulatory factor-6 (IRF6).
(1932-q41) have identified specific mutations in the Mutation analysis demonstrated that popliteal pterygium syndrome can be caused by mutations in syndrome. Many scientists have also reported that the same gene and is therefore allelic to van der Woude VDWS can also be caused due to a microdeletion involving 1q32-q41 in families with this syndrome.
Van Der Woude Syndrome Treatment
- Examination and genetic counseling by a pediatric geneticist (dysmorphologist) should be suggested. Surgical repair of cleft lip and cleft palate or other anomalies may be required.
- Surgical excision of lip pits is often performed for cosmetic reasons even in less severely affected individuals.
Gorlin-Goltz Syndrome
Jarisch and White first reported the Gorlin syndrome in 1894 that was later described in detail by Gorlin in 1960.
Gorlin-Goltz Syndrome Clinical Features
The clinical features that are considered for diagnosing Gorlin syndrome are classified into two groups of criteria; major criteria and minor criteria.
Gorlin-Goltz Syndrome Major Criteria
- Two or more basal cell carcinomas in persons cell growth and division. As a result cells proliferate younger than 20 years.
- Odontogenic keratocysts of the jaw.
- Bifid, fused ribs.
- Calcification of the falx cerebri.
- Palmar or plantar kits.
Gorlin-Goltz Syndrome Minor Criteria
- Syndactyly of digits.
- Macrocephaly.
- Hypertelorism, frontal bossing, cleft lip or palate. is affected with Gorlin syndrome.
- Scoliosis, vertebral anomalies.
Gorlin-Goltz Syndrome Incidence: The prevalence of Gorlin syndrome is estimated to be 1 in 57,000 people.
Gorlin-Goltz Syndrome Genetics
Mutations in the PTCH1 gene cause Gorlin syndrome. This gene encodes for a protein called Patched-1 which functions as a receptor protein. The Patched-1 receptor proteins have specific sites into which certain other proteins called ligands (e.g. Sonic Hedgehog) fit like keys into locks. Ligands and their receptors together trigger signals that affect cell development and function.
Patched-1 prevents cell growth and division PTCH1 gene acts as a tumor suppressor gene which (proliferation) until Sonic Hedgehog is attached. The means it keeps cells from proliferating too rapidly or in an uncontrolled way. Mutations in this gene prevent the production of Patched-1 or lead to the production of an abnormal version of the receptor.
An altered or missing Patched-1 receptor cannot effectively suppress cell growth and division. As a result cells proliferate uncontrollably to form tumors that are characteristic of Gorlin syndrome.
The genetic locus for Gorlin syndrome is located on chromosome bands and subbands 9q22.3-q31. More than 50 germline mutations in PTCH are described. About 40% of cases of Gorlin syndrome represent new mutations in affected individuals.
Inheritance: Gorlin syndrome is inherited in an autosomal dominant trait. Usually one of the parents is affected with Gorlin syndrome.
Waardenburg Syndrome (WS)
It is an inherited disorder often characterized by varying degrees of hearing loss and changes in skin and hair pigmentation.
Waardenburg Syndrome (WS) Clinical Features
- Eyes are of two different colors. One eye is usually brown and the other blue. Sometimes one eye has two different colors.
- Difference in eye color is associated with hearing impairment.
- People with WS may also have distinctive hair coloring such as a patch of white hair or premature gray hair as early as age 12.
- Some individuals may show white patches on the skin.
- Cleft lip and cleft palate may also be associated with WS.
- Some individuals may show intestinal or spinal disorders.
Waardenburg Syndrome (WS) Incidence: About 1 in 10,000 to 1 in 20,000 individuals. mutations in the PAX3 gene. Mutations in the MITF
Types of WS or Types of Waardenburg Syndrome (WS)
Though there are about four different types of Waardenburg syndrome but most common types of WS identified by scientists are Type I and Type II. The type of WS is determined by the distinctive features present in a person.
Type 1 WS-It is represented by persons having wide- set eyes (hypertelorism) due to a prominent, broad nasal root (dystopia canthorum). Hearing impairments occur in about 20% of individuals with this type of Waardenburg syndrome.
Type 2 WS-Persons who do not have a wide set eyes but who have many other WS characteristics are described as having Type II WS. About 50% of persons with WS Type II have a hearing impairment or are deaf. Type II WS can be further classified into 4 different sub types.
Waardenburg Syndrome (WS) Genetics
Inheritance: This condition is usually inherited in an autosomal dominant (AD) pattern. A small percentage of cases result from new mutations in the gene. Some cases of type II Waardenburg syndrome appear to have an autosomal recessive pattern of inheritance. Most often the parents of a child with an autosomal recessive disorder are not affected but are carriers of the disease.
The genes that cause Waardenburg syndrome are involved in the formation and development of several types of cell including pigment-producing cells called melanocytes. Melanocytes make a pigment called melanin that contributes to the coloration of skin, hair, and eye and also plays an essential role in the normal function of the inner ear.
Mutations in any of these genes disrupt the normal development of melanocytes leading to abnormal pigmentation of the skin, hair, and eyes and problems with hearing. In addition to melanocyte development these genes are important for the development of nerve cells in the large intestine.
Mutations in any of these genes result in hearing loss, changes in pigmentation, and intestinal problems Some individuals may show intestinal or spinal related to Hirschsprung disease. disorders.
Type 1 Waardenburg syndrome is caused by and SNAI2 genes are responsible for type 2 Waardenburg syndrome.
Paired box 3 (PAX3) gene is active in cells called neural crest cells. These cells migrate from the Paired box 3 (PAX3) gene is active in cells called developing spinal cord to specific regions in the embryo. The protein made by the PAX3 gene directs the activity of other genes (such as MITF) that signal neural crest cells to form specialized tissues or cell types such as limb muscles, bones in the face and skull (craniofacial bones), some nerve tissue and pigment-producing cells called melanocytes.
Melanocytes produce the pigment melanin, which contributes to hair, eye, and skin color. Melanocytes are also found in certain regions of the brain and inner ear.
Microphthalmia-associated transcription factor (MITF) gene helps to control the development and function of pigment-producing cells called melano- cytes. The official name of this gene is “micro- phthalmia-associated transcription factor” and “MITF’ is the gene’s official symbol. The MITF gene is also known by a few other names listed below. The Snail 2 (SNAI2) gene probably plays a role in the formation and survival of melanocytes.
The mapping analysis by Farrer, et al (1994) indicated that WS type I is linked to PAX gene situated on chromosome number 2; WS IIA is linked to mutation in gene MITF located on short arm of chromosome 3 (3p): WS IIB is due to mutation in gene located on short arm of chromosome 1 (1p) and WS IIC is due to mutation in gene situated on short arm of chromosome number 8 (8p).
Waardenburg Syndrome (WS) Treatment
No effective treatment is available for persons with Waardenburg syndrome.
Osteogenesis Imperfecta
The osteogenesis imperfecta (OI) is a serious inherited disorder. The bones formed are defective, i.e. they are brittle and easily fractured. This condition results due to an abnormality in the formation of type 1 collagen. It is formed either in less quantity or poor quality. 90% of body collagen is type 1. This type of collagen fibers is found in bones, capsule of organs, cornea, dentin, sclera, tendon, dermis, fascia, the dura and ligaments.
Incidence: the disease is observed in 7 per 100,000 persons. Type 2 and 4 are more common than the rest.
The disorder is classified in various types, i.e. OI type 1 to OI type V8. The genetic cause of OI type 5 and 6 are not known. The types 1 to 4 are inherited as autosomal dominant and type 7 and 8 as autosomal recessive traits. Clinical features of only first four types of OI are described in brief:
OI, type 1-It is the mildest form and may remain undetected till the occurrence of the first fracture in a child. The collagen formed in this type of OI is of normal quality but produced in insufficient quantity resulting in the bones getting fractured easily and the spine showing slight curvature defects. Joints are loose or sub-laxed.
The sclera looks blue (sometimes purple or gray) instead of white as the sclera is very thin and the choroidal veins present beneath the sclera gives a blue look to the sclera. Sometimes Type 1 OI is associated with DGI characterized by opalescent teeth.
OI, type 2-This is also known as congenital OI. It is the most severe form of OI and it is lethal at birth or shortly thereafter. As most cases die within first few Mutation of COL1A1 Gene years, this type of OI may not be encountered in dental practice. The collagen produced in neither of good quality nor of sufficient quantity. This leads to severe bone deformity and small stature in persons who survive the disease.
OI, type 3 – This type shows progressive deformities with increasing age, i.e. neonates present with mild symptoms that bloom into severe symptoms with age. The collagen produced is of sufficient quantity but of poor quality. Bone deformities are severe and bone gets fractured easily. Joints are loose. Sclera shows discoloration.
OI, type 4-Collagen produced is of sufficient quantity but poor quality. Affected person is of short stature with deformed and fractured bones and spinal curve disorders. There may be early loss of hearing.
Genetics of Ol or Osteogenesis Imperfecta
The formation of collagen begins as procollagen molecules. Each rope-like procollagen molecule is made up of three chains, the two proalpha 1 chains and one proalpha 2 chain.
The proalpha 1 chain is produced by gene COL1A1 (Collagen type 1 alpha 1) and proalpha 2 chain is produced by gene COL1A2 (collagen type 1 alpha 2) (Byers, et al 1991 and Byers, 1993). Gene COL1A1 is located on the long arm of chromosome number17 between 21.3 and 22.1. Gene COL1A2 is located at 7q22.1.
Two other genes are also involved in the synthesis of collagen type I, i.e. CRTAP (Cartilage associated Two other genes are also involved in the synthesis protein) and LEPRE 1 (Leucine proline-enriched proteoglycan 1). The CRTAP gene codes instruction for making a protein called cartilage associated protein. This protein is critical for normal folding and assembly of collagen. It plays an important role in bone development.
This gene is located on short arm of chromosome number 3 at position 22.3. The LEPRE1 gene is also known as P3H1. It provides the code for making an enzyme called prolyl-3 hydroxylase 1. This enzyme modifies an amino acid called proline in collagen molecules. Proline is necessary for proper folding and assembly of collagen. This gene is located on the short arm of chromosome number 1 at position 4.1.
Mutation of COL1A1 Gene
The mild mutation of this gene leads to OI type 1. There is a reduction in the production of proalpha 1 chain thus reducing the amount of collagen 1 produced. A severe mutation of this gene leads to production of type 2 to type 4 varieties of OI. Deletion of segments of DNA from COL1A1 gene results in nonfunctional proalpha1 chain.
Some mutations may replace the amino acid glycine with some other amino acid. Sometimes mutation may also interfere with the assembly of collagen molecules. Thus the mutation on gene COL1A1 leads to the formation of abnormal types 1 collagen.
Mutation of COL1A2 Gene
This gene codes for proalpha 2 chain of collagen molecule. The mutation of COL1A2 gene produces abnormal collage type 1. The mutation of this gene leads to severe types of OI (type 2, type 3 and type 4).
Mutation of CRTAP Gene
Mutation of this gene leads to type 7 variant of OI. Cartilage associate protein is not formed due to the underlying mutation. This results in the production of abnormal cartilage.
Mutation of LEPRE 1 Gene
The mutation leads to abnormal production of enzyme prolyl-3 hydroxylase 1 which ultimately leads to incorrect folding and assembly of collagen molecules. The mutation of this gene is associated with OI type 8.
Osteogenesis Imperfecta Modes of inheritance-Type 1 to type 4 OI are inherited as autosomal dominant entities while type 7 and 8 are inherited as autosomal recessive ones.
Osteogenesis Imperfecta Treatment
No genetic cure is available at present. Putting metal rods in these bones can prevent fractures of long bones.