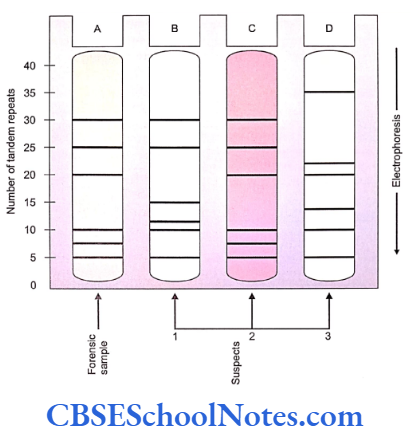
Introduction And Mendel’s Laws Of Inheritance
Definition Of Genetics
We all have observed that children of same parents, more often than not, resemble each other as well as resemble their parents. We also try to guess the proximity of this resemblance towards one of the two parents. It is quite interesting to observe that the resemblances are not just confined to their physical appearances (facial features, height, color of skin and hair, etc.), but are often perceptible in their mental attributes (intelligence, tastes, attitudes, etc.) also.
This is because the characteristics of parents are passed on to the children through the gametes furnished by each parent (sperm an dovum). The process of transmission of characters from one generation to the next (parents to children) is called inheritance or heredity.
The question that crops in our mind is about what are the substrates that actually determine the characters in an individual. The characters, in fact, are determined by certain factors called genes; the fundamental units of inheritance. For details about genes refer to Chapter 4. The genes determining specific characters in an individual are transmitted to them physically through gametes of the parents.
The individual in turn passes these trains onto its offsprings through his or her gametes. An individual is either short or tall in stature or with black or blond hair entirely due to the presence or absence of specific genes responsible for a particular character or trait. Since an individual receives genes from parents (through sperm and ovum), he or she inherits characters both from the father and the mother.
An important fact about gene transmission is that when they are transmitted from one generation to the next, the transmission of a trait is not random but it follows some discreet statistical laws depending upon the type of the character and of course, the behavior of the gene during gamete formation. Therefore the science of genetics can be defined as the study of genes and of the principles that govern the passage of genes from one generation to the next.
Read and Learn More Genetics in Dentistry Notes
Divisions Of Genetics
Human genetics can now be divided into several branches. Few important subdivisions of genetics are as under.
Molecular genetics: Includes the study of chemical structure of gene at molecular level. This branch also includes the study of function of gene and regulation of its activity.
Cytogenetic: Deals with the study of chromosomes. Cytogenetics provides the cytological explanation of different genetic principles.
Biochemical genetics: Concerns with the study of genes and their products, the enzymes, which control important stages of various metabolic processes. This branch deals with the inborn errors of metabolism.
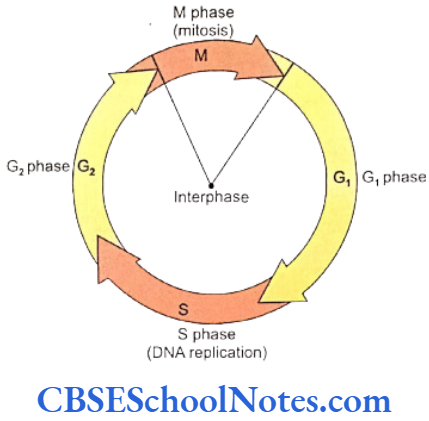
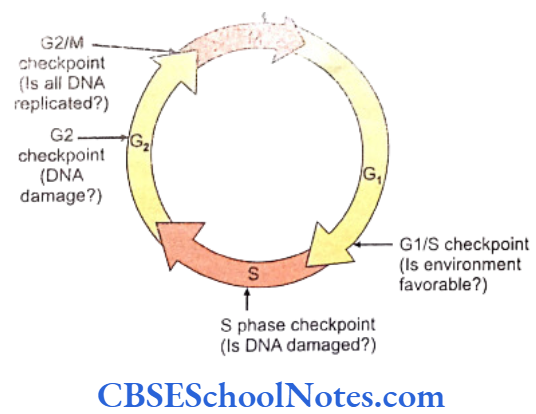
Cancer genetics: The cell cycle is under genetic surveillance and control. The cycle progresses from one stage to the next through several stages called the checkpoints. The structure of all genes is scrutinized at these periodic intervals for allowing only the healthy genes to proceed to the next stage. Cancer genetics studies the abnormalities related to these checkpoints to find the reasons that cause cancer.
Immunogenetics: The immunological make-up of an individual is under strict control of certain genes. Immunogenetics deals with the genetics of production of different types of antibodies.
Developmental genetics: Deals with the genetic control of development of an embryo.
Population genetics: This branch deals with frequencies and distribution of genes in human population and the rates of their mutation.
Classification Of Genetic Diseases
Diverse genetic mechanisms are involved in different hereditary diseases. The cause of a genetic disorder may have its base in the abnormality of the structure of a single gene or multiple genes. Genetic diseases may be also due to a gross abnormality in the structure of an entire chromosome. Thus genetic diseases may be classified as under:
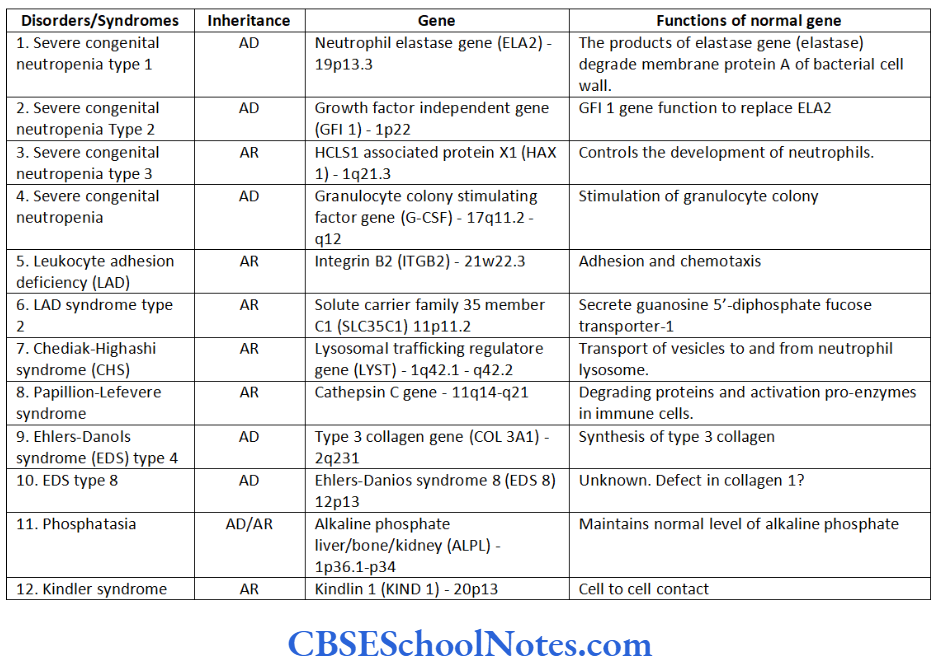
- Disorders due to mutation in single gene: Single gene mutations are responsible for these disorders and they follow laws of Mendelian inheritance. These disorders may be autosomal dominant, autosomal recessive or X-linked. Thousands of disorders can be categorized in this group. Some examples related to dentistry are given below.
- Autosomal Dominant
- Achondroplasia
- Dentinogenesis imperfecta type 1
- Amelogenesis imperfecta hypoplastic type 2 (AIH2)
- Amelogenesis imperfecta hypocalcification type
- Hypodontia
- Osteogenesis imperfecta.
- Autosomal Recessive
- Cystic fibrosis
- Amelogenesis imperfecta (local hypoplastic type)
- Amelogenesis imperfecta (pigmented hypomaturation type)
- Neonatal osseous displasia 1.
- X-Linked Dominant
- Amelogenesis imperfecta (Hypoplastic)
- Vit. D resistant rickets.
- X-Linked Recessive
- Hemophilia
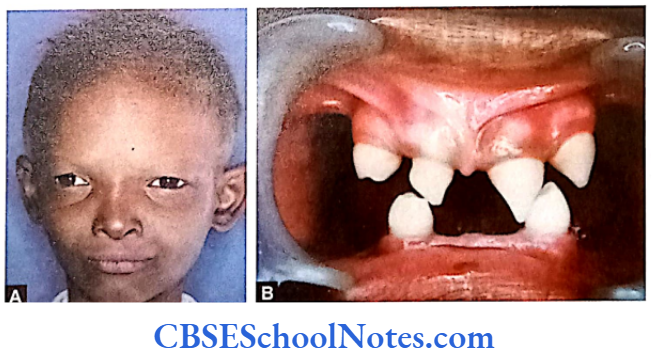
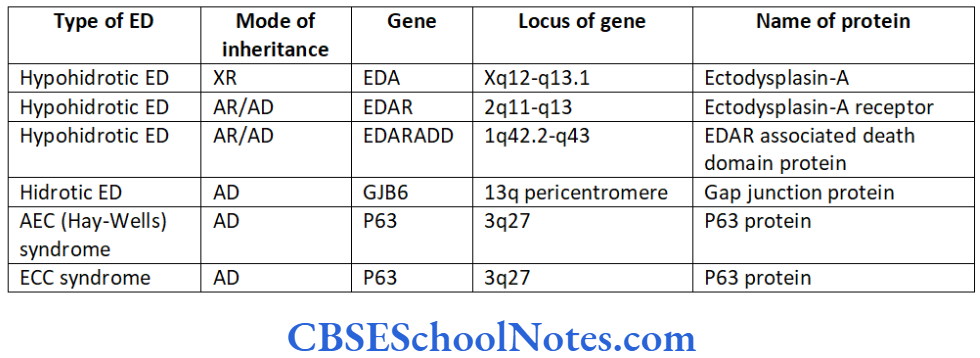
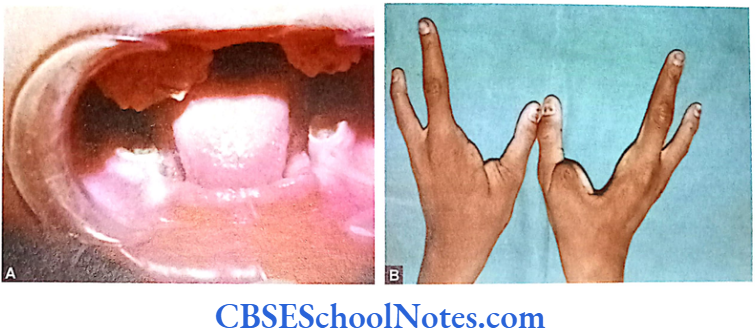
- Ectodermal dysplasia type 4
- Amelogenesis imperfecta hypomaturation type (AIH)
- Chondrodysplasia punctata – 1
- Autosomal Dominant
- Multifactorial disorders: Cumulative or additive effects of multiple genes are implicated in these disorders. The normal characters like height, color of skin, intellignece and physique are determined by the interaction of many genes. Common congenital malformations like cleft lip and palate and diseases like hypertension and diabetes mellitus ae multifactorial disorders. Some kind of oral conditions like dental caries, periodontitis and malocclusion have strong genetic susceptibility. These kinds of disorders are results of interplay between gene expression and environmental factors.
The multifactorial disorders follow different pattern of inheritance as compared to single gene disorders. - Disorders due to chromosomal abnormality: This group includes gross structural anomalies that give rise to alterations in the number of chromosomes (absence of a chromosome or presence of an extra chromosome), i.e., Trisomy 21 (Down’s syndrome) or Turner’s syndrome (XO). This class also includes disorders, which result due to abnormality in the structure of chromosomes such as deletions and translocations. The invention of banding and FISH (fluorescent in sity hybridization) techniques has helped to detect even minor abnormalities in chromosomes. Subtle or point chromosomal abnormalities are included in the single gene disorders.
- Somatic genetic disease: Cell divisions (mitosis in somatic and meiosis in germ cells) constantly occur during the life time of an individual. During each cell division there are chances that a change in the structure of a gene (gene mutation) may take place due to an error in DNA replication. It may also happen that at the end of a cell division (mitosis or meiosis) one of the daughter cells might receive an unequal number of chromosomal (due to erro in chromosomal separation). These kinds of mutation or mistakes in chromosomal distribution are accountable for numerous somatic and germ line diseases.
Genetics In Dentistry
It was first observed by the French biologist Maupertius (1689-1759) that the conditions like polydactyly and albinism were inherited in human beings. Likewise John Dalton (1766-1844) observed that color blindness and hemophilia were inherited diseases. However, human genetics was recognized as a science only after rediscovery of Mendel’s Laws of Inheritance in early 1900.
From the mid 20th century onwards, oral health care professionals had started realizing that many diseases related to the oral cavity were in fact inheritable. Information from the Human Genome Project (2001) and recent genetic researches has clearly indicated that many dieseases with the dental, oral and craniofacial manifestations have a genetic basis both in terms of heritability (disease running in families) as well as arising from structural mutation in a particular individual.
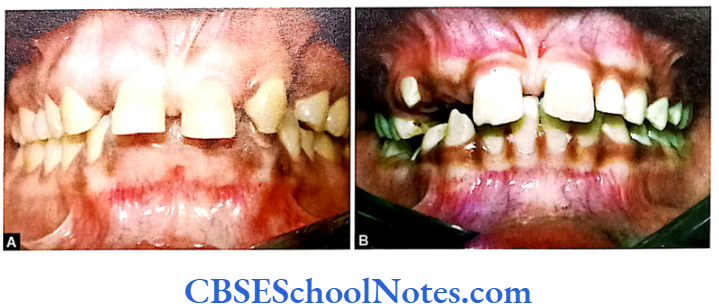
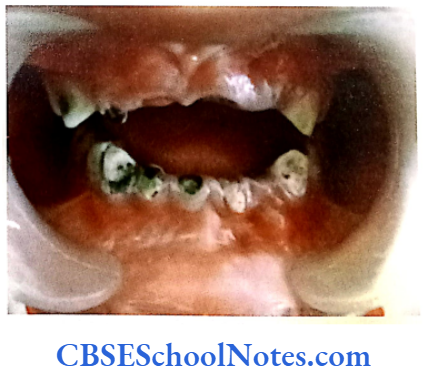
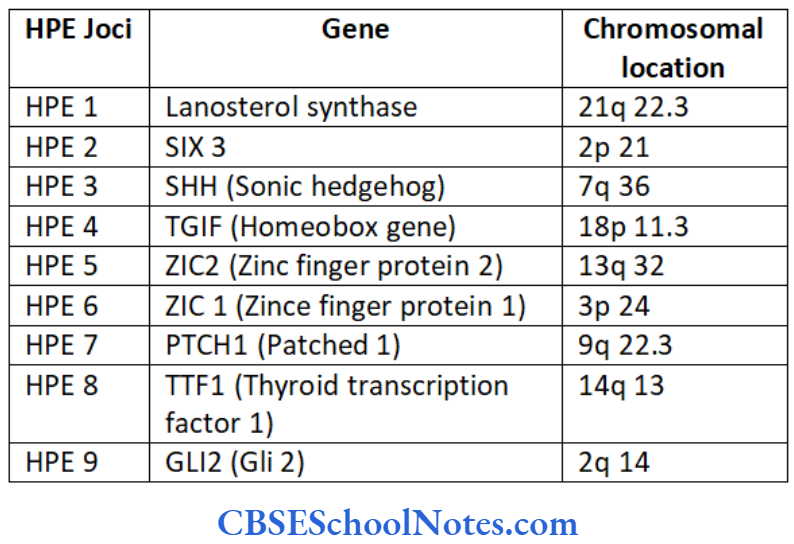
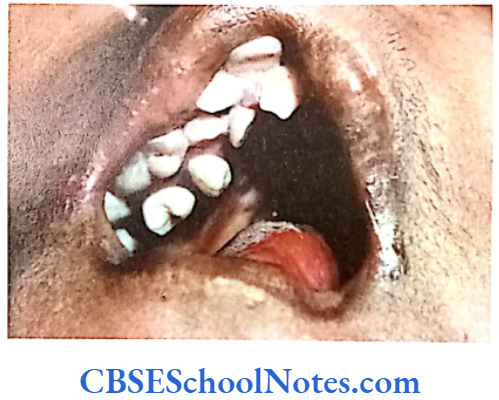
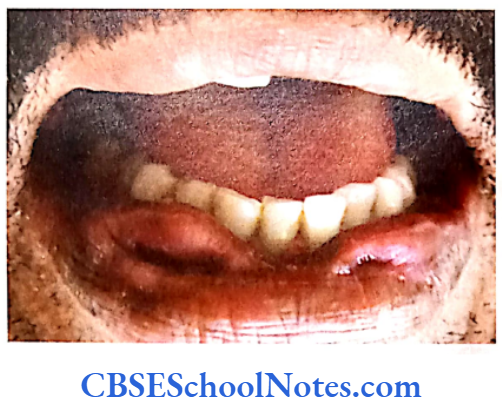
Tooth Agenesis
The etiology of tooth agenesis was largely unknown till the recent past. But today we know that the development of tooth is strictly under the control of many genes. Several mutations in the developmental genes could result into failure of tooth development. Familial tooth agenesis may be transmitted as an autosomal dominant, recessive or an X-linked condition.
Similarly, most cases of hypodontia exhibit polygenic inheritance pattern. Hypodontia is associated with syndromes like Down’s syndrome, ectodermal dysplasia and the Ellis-van Creveld syndroms. This demonstrates that the development of other organs and tissues of the body is closely related to the development of dentition and perhaps regulated by common genes.
Dental Caries
Certain microorganisms have been incriminated as the causal factors for two major diseases of oral cavity, i.e. dental caries and periodontal diseases. Recent research date have pointed that these conditions have a strong genetic predisposition. Different people have different susceptibility risk for developing periodontics.
Studies have shown that the increase vulnerability to severe adult periodontics is due to variation in the interlukin-1 (IL – 1) gene cluster that is situated on chromosome number 2.
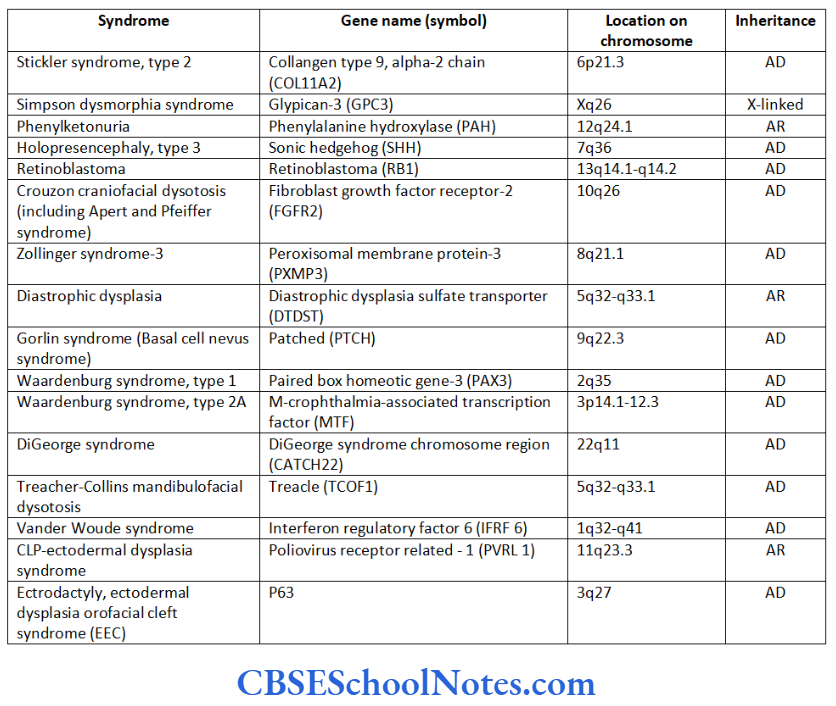
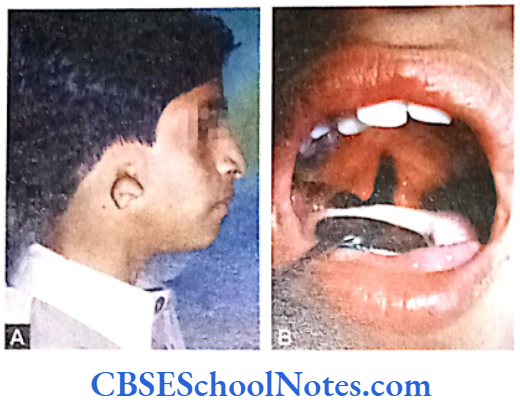
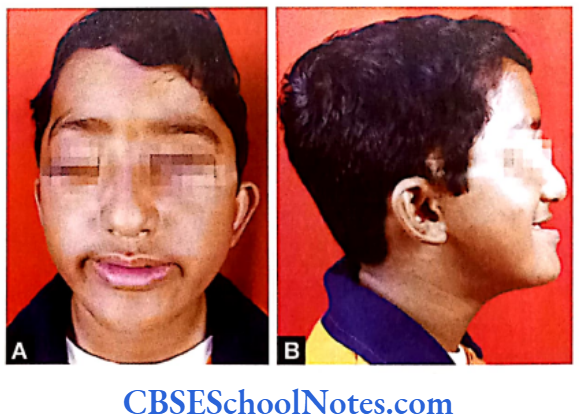
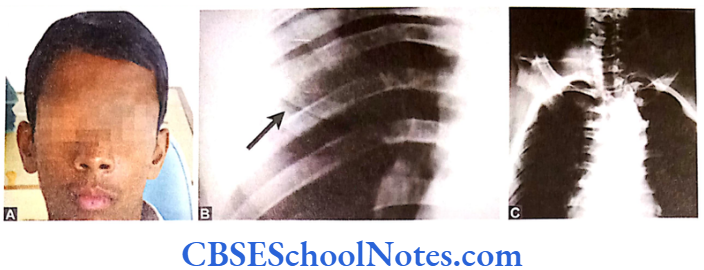
Craniofacial Syndromes
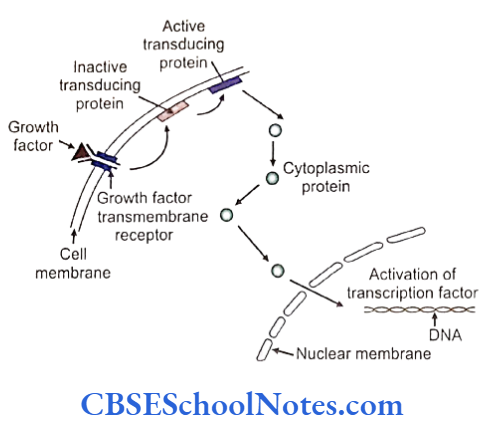
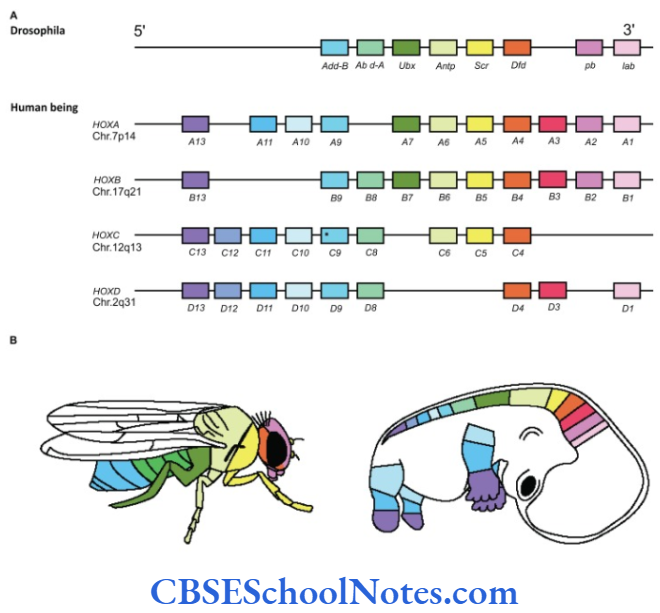
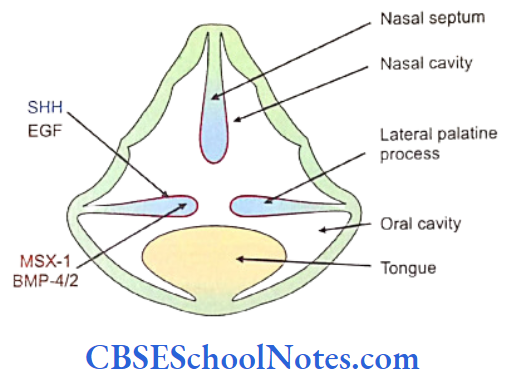
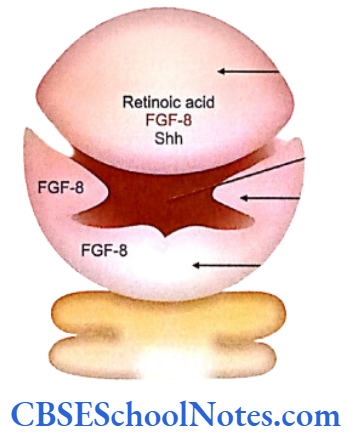
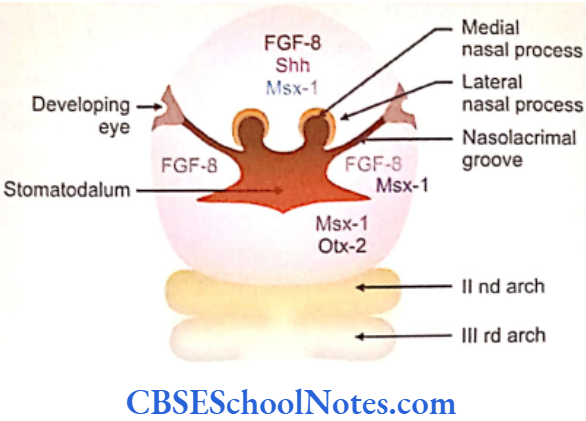
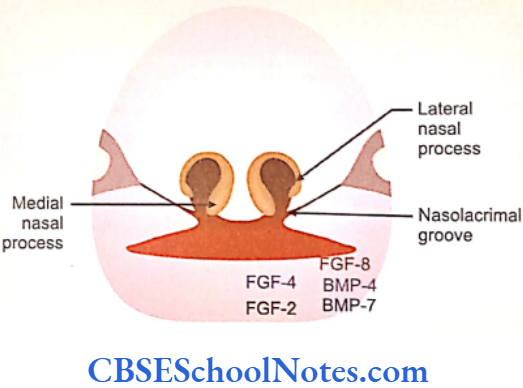
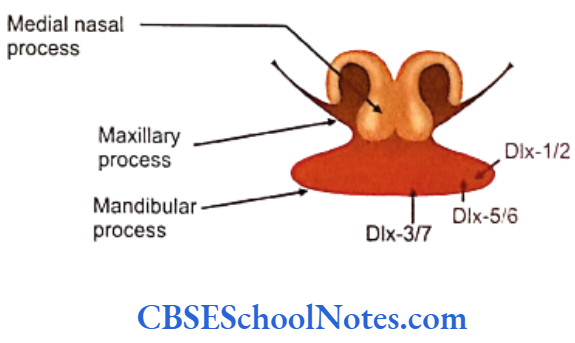
The development of craniofacial region during the early stages of development is genetically determined in terms of migration of definite neural crest cell and through this to the expression of certain sequential homeobox genes. Epithelial-mesenchymal interaction during embryogenesis is regulated by growth factors and the retinoic acid super families.
Conditions like hemifacial microsomia and craniosynostosis have their origin in neural crest cell disorders. The nutation in fibroflast growth factor receptor genes are responsible for abnormal suture development and found to occur in Apert, Crouzon and Pfeiffer syndromes. Cleidocranial dysplasia is characterized by defects in the membranous bones of the cranial vault and clavicle.
The nutations responsible for this deformity have been found to occur in the core binding factor 1 gene (CBFAI). The gene responsible for a well known craniofacial abnormality the Treacher Collins syndrome is situated on the long arm of chromosome 5. Many craniofacial abnormalities are due to interaction between environmental and genetic factors.
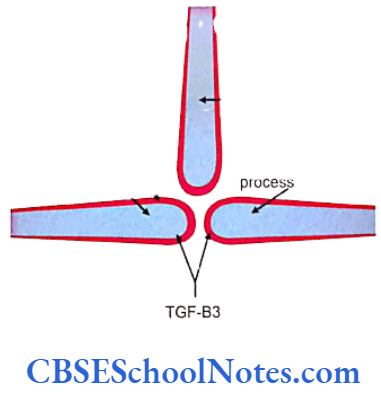
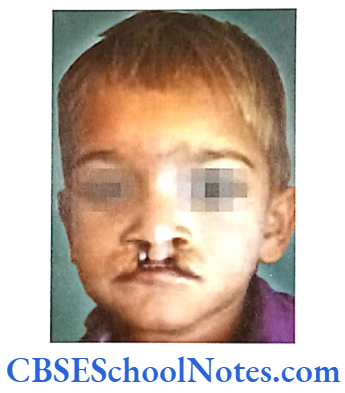
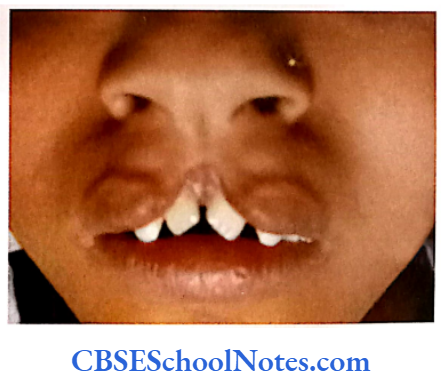
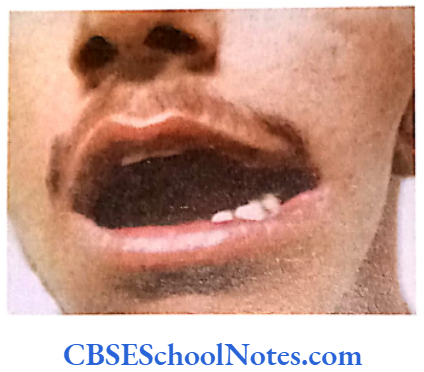
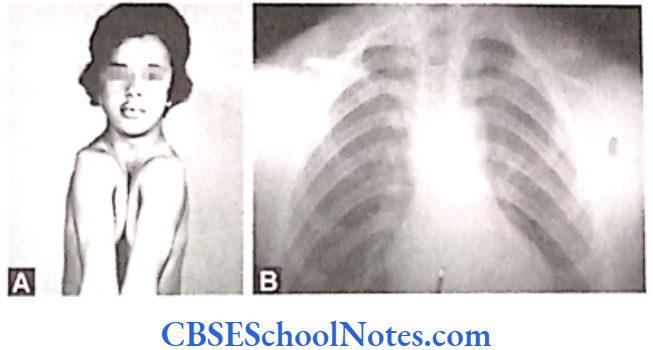
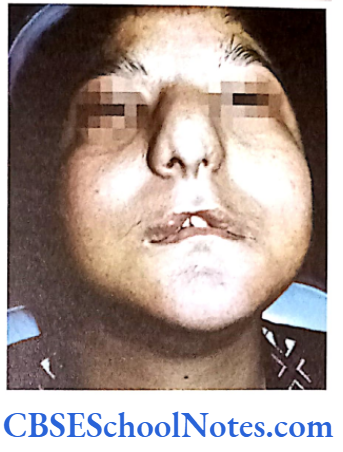
Cleft Lip, Cleft Palate and Cancers
Among the common occurring malformations of the oral cavity Cleft lip and cleft palate are amongst the top in the list. These congenital malformations are inherited as multifactorial traits. The same is true for malocclusion.
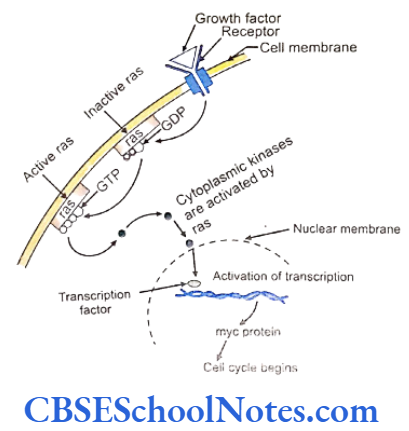
The head and neck region are very common sites for carcinomas in general and oral cancer are the ones seen quite frequently. The dynamics of cancer involves changes in the genome that result in uncontrolled cellular proliferation and metastasis.
The growth factor and growth factor receptor genes regulate the proliferation of cells. Genes responsible for cancer are known as oncogenes. These genes function normally in regulating cellular activity. A mutation in these genes may trigger them to acquired oncogenic properties. Cell division is strictly under genetic control and each of the steps is under constant surveillance of cellular mechanisms.
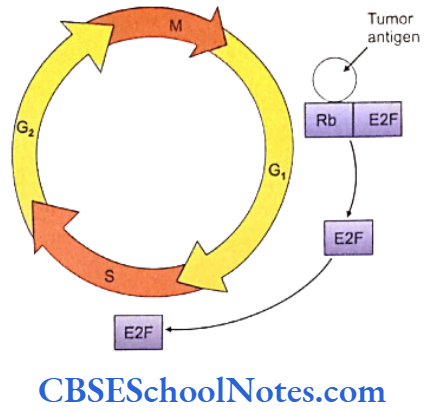
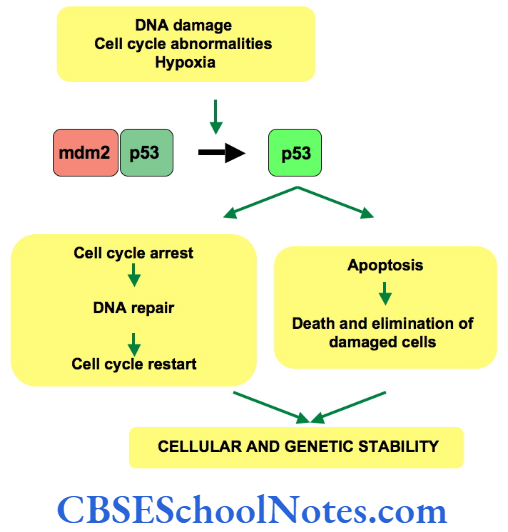
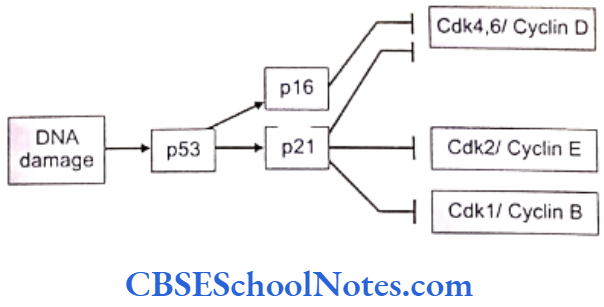
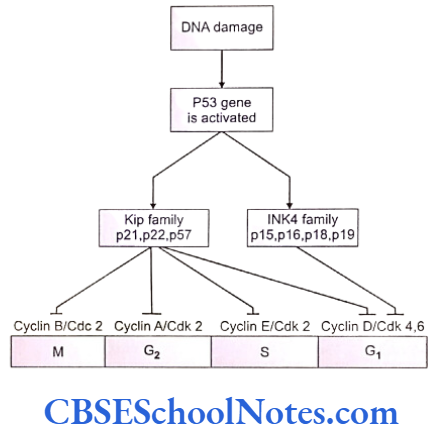
Cell cycle checkpoints exist at appropriate transition points of the cell cycle. The activites at these checkpoints are executed by special proteins that are synthesized by specific genes like the p54 gene. Anomalies in these genes lead to abnormal cell division and subsequently to tumor formation. Structural integrity of the DNA us determined and checked at the checkpoints before allowing it to proceed to subsequent stages of cell division.
Mutations of the check point controlling genes and proteins cause several cancers. The tumor suppressor genes constitute another important cell cycle controlling element. These genes apply brakes to the events in a cell division in case of detection of an abnormality at any stage. These genes are constitutively active or in simple terms, active by default in all normal cells. A mutation causing abnormal activity of any tumor supressor gene may lead to cancer.
Researchers have also identified several tumor-forming genes that occur in normal cells but remain inactive by themselves. Such proto-oncogensea trigger unwarranted cell division if they are activated by any means or when any normally occurring inhibition acting on them is withdrawn .
It is hopeted that the near future will witness a lot of excfiting advances in: Use of primary teeth as source of stem cells, tissues engineering in dentistry, use of saliva as a diagnostic fluid in deteching genetic dental disorders and salivary gland gene transfer.
It is quite imperative that dental practitioners now will increasingly require knowledge of human genetics and the awareness of the applications of new molecular based diagnostic and therapeutic technologies. Thus a sound knowledge of genetics will definitely improve the ability of dentists to diagnose and treat patients suffering from inherited and genetically caused dental diseases.
Since recent past more and more diseases are being recognized as having something related to genes and genetics. This is perhaps due to interpretations based on our new and expanding knowledge at the molecular level and progress in modern diagnostic techniques.
On other hand this may also be due to the fact that owing to the overall improvement in hygiene and health care, the incidence of communicable diseases and nutritional deficiency has reduced thereby shifting our attention to diseases resulting from gene-related etiology. Genetic disorders are now considered significant causes for disease in all age groups.
Mendel And His Laws Of Inheritance
Johann Gregor Mendel was born in Austria on July 22, 1822. He had to face relentless difficulties in his childhood and youth due to poverty and ill health. It was to the credit of his young man that he remained steadfast in the face of all the adversities in the face of all the adversities for the pursuit of knowledge. It took him almost eight years to complete his initial experiments on pea plants.
Mendel published his reports in the proceedings of the Brunn Natural Science Society in 1866. Mendel’s work remained unappreciated and unnoticed till the turn of the century when the postulates of Mendel were rediscovered and revisited by three independently working scientists, Erich Von Tshermak, Hugo de Varies and Carl Correns in the beginning of the 20th century.

Mendel’s work did not get recognition during his lifetime. He passed away in 1844, much before his monumental work immortalized him as the ‘father of modern genetics’.
The Pea Plants Experiments
Mendel’s experiments were designed to find out the mechanisms responsible of inheritance of traits in the peas plants. His experiments basically involved two types of crosses. One between plants differing in a single pair of contrasting characters such as cross between a pure tall and a pure dwarf plant called the Monohybrid cross and subsequent crosses within the offsprings in each generation obtained from the monohybrid crosses.
The other type of experiment called the Dihybrid cross was carried out between plants differing in two pairs of contrasting characters; a cross, for example, done between plants having yellow and round seeds and plants having green and wrinkled seeds. The contrasting pairs of characters in the dihybrid cross were represented by the color and the texture of the seeds in the two different varieties of plants.
Explaining Certain Terms
Self-pollination: Polines of a flower pollinating the stigma of the same flower is called as self-pollination (self-fertilization).
Cross-pollination: Pollens of a flower pollinating different flower stigma is called cross-pollination. (cross-fertilization). The offspring which result from cross breeding between pure strains is called a hybrid.
Monohybrid cross: The cross between the plants or animals differing in single pair of contrasting characters is called monohybrid cross, e.g. cross between tall and dwarf plants.
Dihybrid cross: The cross between plants or animals differing in two pairs of contrasting characters is called dihybrid cross, e.g. plants with yellow and round seeds are crossed with plants having green and wrinkled seeds.
Monohybrid Crosses Experiments
Mendel crossed plants that differed only in a single pair of contrasting character or trait. He crossed between pure tall plants and pure dwarf plants. The character or trait in case of these plants was the height of the stem and the pair of contrast was the tallness and dwarfness of the respective plants. The purity was verified with repeated self-pollination where the tall plants always inbreeded tall and the dwarf plants yielded dwarf offsprings on repeated inbreeding for several generations.
And when these two varieties of plants (tall and short) were crossed, Mendel observed that:
- All the hybrid members of the first generation, called the First filial generation (F1), were tall plants.
- When the F1 population was allowed to self-pollinate, the palnts of the F1 generation gave rise to the Second filial generation (F2) with the following features.
- The character of dwarfness that had disappeared in the F1 reappeared in F2.
- 75% of the F2 plants were tall and 25% were dwarfs.
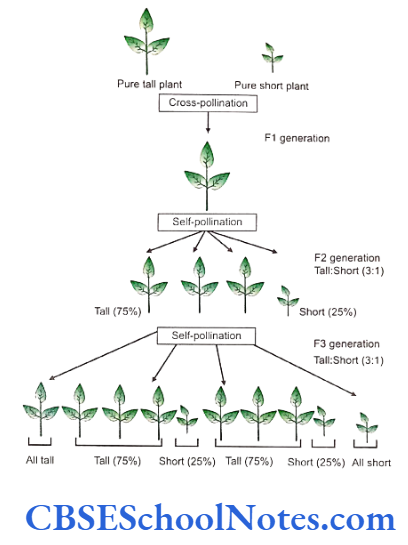
- After self-pollination the F2 generation gave the following results:
- The dwarf plants of F2 when pollinated with dwarfs in the F2, always yielded dwarfs.
- The tall plants when pollinated with the other tall plants within the F2, olny 1/3rd of plants always yielded tall offsprings.
- Remaining 2/3rd of the tall plants yielded tall and dwarf plants in the ratio of 3:1.
Similar to the experiments in monohybrid crosses related to the height of the stem of plants, Mendel conducted experiments with other contrasting characters such as teh shape and color of the seeds and pods, etc. and remarkably got statistically comparable results.
Mendel was not only able to put forward the principles of heredity; he could also predict many of the outcomes of his experiments. He derived several conclusions related to the governance of hereditary traits that hold well till today.
- Inheritable characters are transferred with the help of factors through generations. These factors were later identified as genes.
- The heritable factors are transmitted through gametes (sperms and ova).
- The factors for each character or trait exist as a pair.
Each of the factors (genes, as we know them today) is responsible for a trait and is located at identical positions on each chromosome of a particular pair of chromosomes (homologous chromosomes). The fixed position on a particular chromosome for a definite gene is called the locus (plural loci) for the gene.
The same positions (loci) on two homologous chromosomes contain genes responsible for the same character. For example the loci representing the height of a plant may contain genes responsible for tallness (T) or dwarfness (t) in different combinations as (TT, Tt or tt). The alternative form of genes, e.g. ‘T’ and ‘t’ present at the same locus are called allelomorphs or alleles.
The alleles define a particular character depending upon their dominance with respect to each other. Homolous chromosomes carrying identical alleles (same gene) are termed homozygous. A situation with different genes at the loci defining a particualr trait is called heterozygous. - The members of the homologous pair of chromosomes separate from each other at the time of gametogenesis.
Each of the gametes carries only a single chromosome out of the homologous pair. A single gamate will carry a single locus containing either a dominant or a recessive gene depending on the chromosomal constitution of the individual parent.
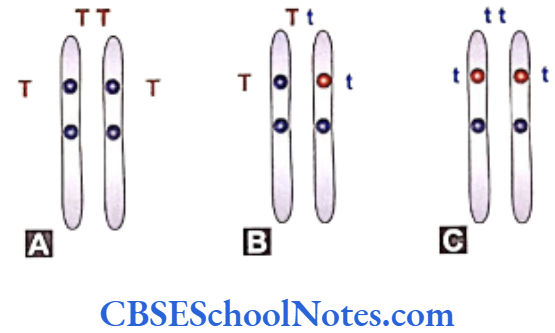
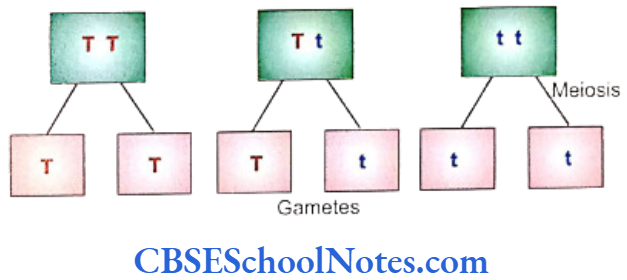
- Crossing between plants of pure variety differing in a single pair of contrasting character yields only the dominant character in first generation, whereas both the characters are expressed in second generation.
Punnett squares are grids that are extensively used to compute the genetic constitution of an individual by entering the constitution of the gametes on the top and side squares of the grid. Analyzing the above shown cross that is same as the first cross in Mendel’s monohybrid cross experiment to yield the first filial generation in the Punnett square, looks like as below.
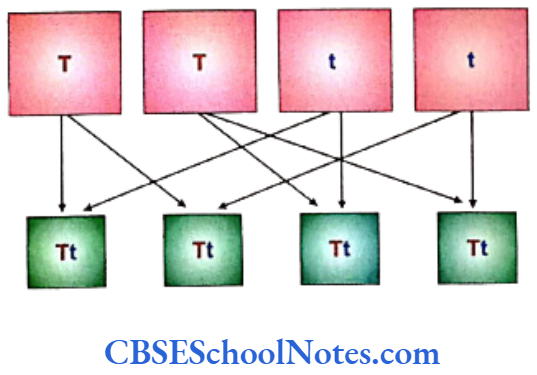
The genotype of an individual is defined as its genetic constitution for any particular trait. The term phenotype denotes the physical appearance for a particular trait. A gene is considered to be a dominant if it is able to express itself in the phenotype even when it is present in a heterozygous condition, e.g. the gene for tallness ‘T’ in (Tt). The gene for shortness ‘t’ is only expressed when it is present in a homozygous state (tt) in an individual and is called a recessive gene.
It is thus evident that all the progeny tall plants of the F1 generation (Tt) have different genotypes than the pure bred parent tall plants (TT) though they are same in their phenotypes.

Self-pollination of F1 plants can be analyzed in the F2 generation with the Punnett square.
Self-pollination in the F1 generation yielded the F2 progeny. The F2 progeny constituted two varieties of phenotype with the reappearance of short stature in the plants. The genotype of the tall plants showed two varieties; the homozygous tall the heterozygous tall plants. The concepts of dominance and recessive perspectives are also clear from the results in the grid squares.
Characters are transmitted from one generation to next following statistical laws. When the plants of F1 generation were self-pollinated both tall and short plants appeared int he ratio of 3:1. When the plants of F2 generation were self-pollinated the tall and short plants always appeared in the fixed ratio.
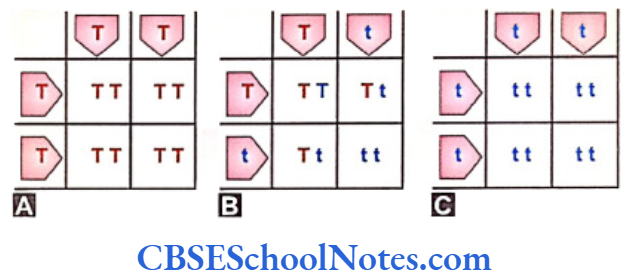
The results of the F3 generation in the Punnett square show the genotype and the phenotype of the individual progeny.
Dihybrid Crosses Experiments
For his dihybrid cross experiments Mendel selected two varieties of pea plants that differed in two pairs of contrasting characters. He selected, for example, pure variety of plants having yellow and rounded seeds and crossed them with another pure variety of plants having green and wrinkled seeds (dihybrid cross).
These crosses were conducted to study the inheritance of a pair of contrasting characters with relation oto the inheritance of the other coexisting pair of contrasting character in successive generations.
Mendel found that in F1 generation all plants were yellow with round seeds indicating that the yellow color and the round shape as were dominant over the green color and wrinkled shape that were recessive in nature.
On self-pollination, the F1 plants yielded the F2 progeny. These offsprings were of four different phenotypes in the ration 9:3:3:1 with 9 yellow and round, 3 yellow and wrinkled, 3 green and round and 1 green and wrinkled types of seeds.
It was thus observed that the two pairs of contrasting characters actually were transmitted independent of each other. The offsprings even demonstrated the new combination of characters in the form of yellow and wrinkled and green and round seeds in the F2 generation. The details of the experiments and their discussion are not discussed here.
Carl Correns, one of the rediscoverers of Mendel’s work in 1900, promoted the ideas of Mendel as the “laws of inheritance”. Following three concepts are recognized as Mendel’s Laws.
Mendel’s Laws
It was earlier believed that traits or characters of parents become blended, diluted and lost in the offsprings of subsequent generations. Mendel’s experiments have shown that these parental characters are determining by certain ‘factors’ (genes) and do not “mix” ir “contaminate each other” and expressin the progeny at a later stage. Mendel’s first law of inheritance was based on this evidence.
The Law of Uniformity
Plants with two contrating (one tall and the other short) characters when crossed, the characters do not blend. If any character is not expressed in the girst generation it may reappear without change in a subsequent generation.
The Law of Segregation
An individual possesses two factors (genes) for a particular character with each of these factors situated on one of the chromosomes of a homologous pair. At the time of formation of gametes each member of the pair of chromosome separate independently from one another so that each gamete carries only one chromosome of the pair and as such only one of the two factors (gene) responsible for the determination of a character.
In Mendel’s words, neither of the factors has “taken over anything from the other”. The genes of a pair are separated completely unaltered on a chromosome that migrates to a gamete during gametogenesis.
The Law of Independent Assortment
Members of different gene pairs (determining different sets of characters) that exist on the same chromosome, assort independent of each other during gametogenesis to migrate into a gamete. Because of such independent assortment new combinations between different sets of characters are produced in an offspring. For explanation refer to dihybrid cross.
The dihybird cross experiments yielded four different phenotypes in the F2 generation with yellow-round, yellow-wrinkled, green-round and green-wrinkled seeds. This implied that the genes responsible for yellow and green colors and round and wrinkled shapes of seeds separated out independently tha tresulted in four different phenotypes and 9 different genotypes in the F2 generation.
Summary
- Conclusions from hybridization experiments:
- The factors responsible for inheritance of character are basically the genes.
- The factors or genes for each character occur in pair.
- These genes are transmitted from one generation to next through gametes.
- Members of a pair of genes separate from each other at the time of gametogenesis so that each gamete carries only one gene.
- Only one character (dominant) is expressed in first generation and both characters (dominant and recessive) are expressed in second generation when pure bred plants differing in pair of contrasting character is crossed.
- Statistical laws are followed in transmission of characters.
- Inheritance of one pair of factors is independent to other pair of factors in case of dihybrid cross experiments.
- Hybrid: Offspring of cross (mating) between two generically different organisms.
- Monohydrid Crosses: Cross (mating) between individuals or plants differing in a single pair of contrasting characters. Such cross yields monohybrids which are genetically heterozygous for the particular trait and factor.
- Dihybrid crosses: Cross (mating) between individuals or plants differing in two pairs of contrasting characters.
- Locus: The position of a gene on a chromosome is called locus.
- Allele: Alternative form of a gene present at any particular locus.
- Homologous: Chromosomes come in pairs in autosomes and as sex-chromosomes in the female. The members of the pair are identical to each other in their morphology. These chromosomes of a pair are called homologous.
- Homozygous: A condition of having same allele at a given loci on homologous pair of chromosomes.
- Heterozygous: A condition of having different alleles at a given loci on a homologous pair of chromosomes.
- Genotype: The genetic constitution or makeup of an individual.
- Phenotype: It is the physical, mental or biochemical manifestation of an individual in relation to a particular character resulting from the expression of associated genes. Phenotype may be influences by environmental factors.
- Dominant: Is a trait can express itself even in heterozygous state of a particualr gene (single dose) eg. tallenss.
- Recessive: It is a trait which is expressed only in homozygous condition (double dose) e.g. shortness of a gene.
- Mendel’s laws of inheritance:
- The law of uniformity.
- The law of segregation.
- The law of independent assortment.