Genetics Of Periodontitis
Periodontitis is a dental disorder involving inflammation and infection of the ligaments and bones that support the teeth and results from progressive and uncontrolled gingivitis.
Classification of periodontitis is based on the rate of disease progression. Periodontitis can be divided into two major types, e.g. aggressive (localized aggressive and generalized aggressive) and chronic.
Until recently, periodontitis was thought to be strictly determined by environmental factors like poor oral hygiene, smoking, low socioeconomic condition, bacterial infections of oral cavity, etc. alone. It is a common observation that given the same status of poor oral hygiene and sharing the same environmental factors, some people show severe periodontal disease while some of the same people suffer from mild or no disease.
This indicates the existence of individual differences in susceptibility to the disease and perhaps towards a genetic basis for the susceptibility. Scientists are trying to find out the genes responsible for the disease. It is now well known that periodontal disease is multifactorial (complex) and its susceptibility is influenced both by genetic and environmental factors.
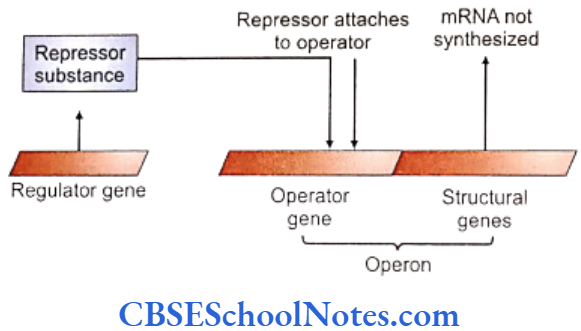
Genes responsible for multifactorial disease are also known as susceptibility genes. However, these genes alone do not produce the disease unless they are exposed to the necessary environmental factors.
Periodontitis is also associated with many types of syndromes and genetic disorders. Periodontitis presents itself as one of the clinical manifestation of the syndrome. This establishes the genetic basis of periodontitis as these syndromes result due to mutation in a single gene/genes.
Read and Learn More Genetics in Dentistry Notes
Verification of the Genetic Basis of Periodontitis
Following genetic analytical methods have been used to evaluate the genetic basis of periodontitis.
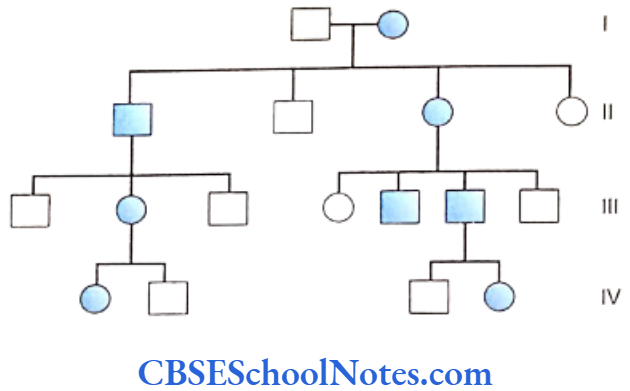
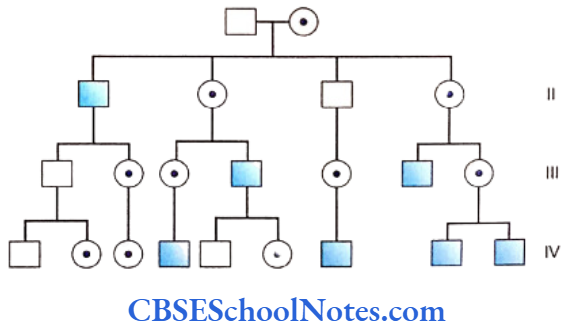
Familial Aggregation Of Periodontal Diseases
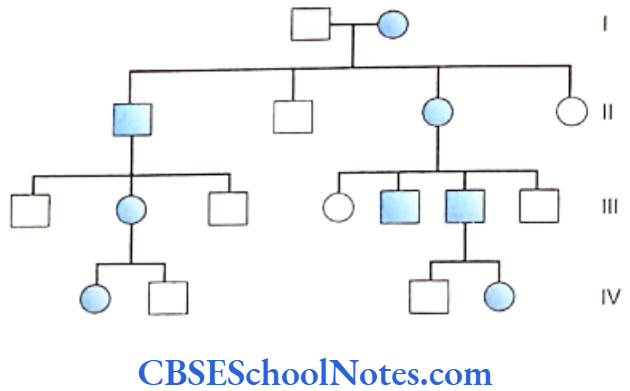
Several members of the same family suffering from periodontitis is indicative of a genetic etiology. Many scientific studies have indicated familial aggregation for both aggressive (Boughman et al., 1988; Long et al, 1987; Marazita et al, 1994 and Hart and Korman, 1997) and chronic periodontitis (Hassell and Harris, 1995). Reports of the familial nature of chronic forms of periodontitis are less frequent.
This aggregation within families strongly suggests of a genetic predisposition to the disease. However, the familial aggregation of the disease may also reflect on the common environmental factors prevalent in the family, i.e. levels of oral hygiene, shared transmission of bacteria, same socioeconomic condition, sanitation, etc.
Therefore familial aggregation is only suggestive of a genetic basis but does not prove a definite genetic etiology. To substantiate evidence in favor of a genetic basis of familial aggregation, application of more specific tools such as segregation and twin studies are imperative.
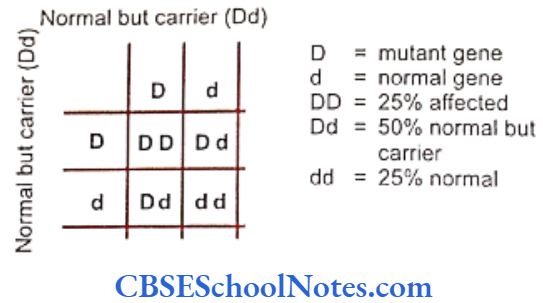
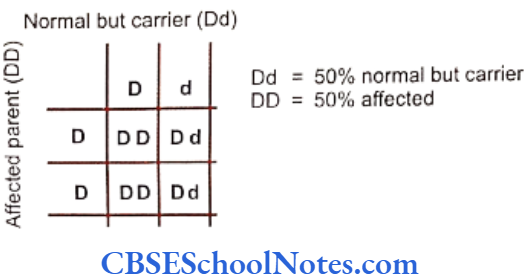
Segregation Analysis
Segregation analysis is conducted in affected families to determine the mode of inheritance of a disease whether it follows an autosomal-dominant, autosomal- recessive, X-linked dominant, X-linked recessive or multifactorial mode of inheritance.
Many studies have been conducted to determine the mode of inheritance of diseases. These studies have indicated many patterns of inheritance in periodontitis. The most definitive segregation analyses in North American families were performed by Marazita and coworkers (1994) who studied more than 100 families segregating aggressive forms of periodontitis and found support for an autosomal-dominant transmission.
They concluded that autosomal-dominant inheritance with approximately 70% penetrance occurred for both Blacks and nonBlacks. It is believed that various forms of aggressive periodontitis are due to single gene defects that are inherited as autosomal- dominant disorders with incomplete penetrance. Some studies have indicated an autosomal recessive mode of inheritance (Saxen, 1980; Saxen and Nevanlinna, 1984). Few workers have also reported X-linked inheritance (Melnick et al, 1976) in the same context.
Why were these studies unable to point towards a single type of mode of inheritance? It might be because of the multifactorial or polygenic nature of the disease or probably due to the fact that the origin of periodontitis is heterogeneous in nature. There is a common precedent in genetics, with relation to heritable pathologic conditions, to show different modes of inheritance in different families. These findings reflect that different genetic loci are capable of causing the same disease in both dominant and recessive manners.
Even though segregation studies have shown different modes of inheritance, they all support the involvement of genes in the causation of the disease.
Twin Studies
Many human traits are multifactorial (polygenic). The modes of inheritance in which can be manipulated with environmental modifications. Twin studies allow evaluation of the importance of genetic as well as environmental factors in causation of a disease. As early as in 1940, Noack recognized similarities of features in periodontal conditions seen in identical twins. Many twin studies in the past were carried out on monozygotic adult twin groups. These twins were grouped as the reared-together and reared-apart sections. Following conclusions were drawn that:
- The periodontal conditions in identical twins were similar.
- A significant genetic influence on various parameters of periodontal diseases was found.
- Findings in the reared-together and reared-apart groups were similar which strongly suggested a genetic basis of the disease. This finding also indicated that environmental factors had no significant influence on development of the disease.
A study by Corey et al (1993) on monozygotic and dizygotic twins indicated a higher concordance rate for monozygotic twins (23% for monozygotic twins as compared to 8% for dizygotic twins). Concordance implied that both the twins of a pair were suffering from periodontitis. This strongly suggested the involvement of a genetic component in the etiology of periodontitis as we know that monozygotic twins possessed identical genes.
Michalowicz et al (1991) investigated alveolar bone height and probing depth in twins from the Minnesota study and showed significant variations in them, according to the differences in their genotype. The twin groups had similar smoking histories and oral hygiene practices. It was concluded that genetics plays a role in moulding the susceptibility to periodontal disease.
In another study Michalowicz (1994) found that monozygotic twins reared either together or apart have been found to bear a more similar type of periodontal disease experience than found between dizygotic twins. These findings indicate that genetic factors may influence the manifestations of periodontal disease.
In a more recent study (Michalowicz et al, 2000) of another large human twin cohort, hereditary factors accounted for approximately 50% of “adult period- ontitis”. The heritable component for periodontitis was not associated or influenced by behaviors such as smoking, utilization of dental care, and oral hygiene habits. This indicated that development of periodontitis was influenced by genes that mediated biological mechanisms.
Twin studies were also used to know about the effect of host genes on the composition of micro- bacteria in the oral cavity. The long-term colonization of bacteria in the oral cavity may be due to the genetic constitution of the host. Studies have revealed that adolescent twins were more similar in their oral microbiota than pairs of unrelated individuals (Moore et al, 1993).
However, the study of Michalowicz et al (1999) in adult twins indicated that neither the host genes nor the environment played a significant influence on the presence of bacteria in subgingival plaque. These two studies suggested that although the host genes influence initial bacterial colonization but this influences do not persist till adulthood.
Linkage Studies For Periodontitis
Linkage studies are conducted to localize the disease causing gene on a specific chromosome. Till date only very few linkage studies analyzing aggressive periodontitis are reported.
- Saxen and Koskimies carried out the first linkage study in 1984 on Finnish families to test the claim for association of aggressive periodontitis with HLA antigens. This study concluded that the gene responsible for aggressive periodontitis was not linked with HLA antigen in these families.
- Boughman et al reported the second linkage study in 1986 for a Maryland population. According to this study the gene responsible for aggressive periodontitis was situated on the long arm of chromosome number 4 (4q 11-13). The inheritance pattern of this gene was determined as autosomal- dominant. The gene was found to cosegregate with the gene responsible for dentinogenesis imperfecta. Both the genes were linked, i.e. present close to each other on the long arm of chromosome number 4.
- However, another study in 1993 (Hart et al.) on a different population of USA failed to locate the gene for aggressive periodontitis on chromosome number 4. This finding indicated that aggressive periodontitis was heterogeneous in nature, i.e. different genes may be responsible for different forms of aggressive periodontitits.
- Prepubertal periodontitis (PPP) is a rare and rapidly progressive disease of young children that results in destruction of the periodontal support of the primary dentition. The condition may occur as part of a recognized syndrome or may occur as an isolated finding. Both autosomal dominant and recessive forms of Mendelian transmission have been reported for PPP. Hart et al. (2000) have localized a gene of major effect for PPP on chromosome 11q14. This PPP candidate interval overlaps the region of chromosome 11q14 that contains the cathepsin C gene responsible for Papillon-Lefèvre and Haim-Munk syndromes.
- Recently in 2004, a linkage study by Li and coworkers mapped the gene responsible for localized aggressive periodontitis on the long arm of chromosome number 1 (1925).
Association Studies For Periodontitis
The susceptibility to a disease depends upon the genetic background of a person. Since periodontitis is an inflammatory disease, it is believed to be affected by genes that are involved in the regulation of inflammatory cell functions. Associations between periodontitis and genes responsible for control of inflammat response are of great value in understanding the genetics of periodontitis.
The immune system controls inflammatory responses against a disease. However, the immune response to a disease varies from person to person and the pattern of immune response in an individual is determined by the genetic make-up unique to the person. The clinical presentation of a disease, therefore, varies in different persons reflecting the diversity of immune response shown by each individual. The same concept is also applicable to developing specific and targeted medical treatment.
Thus if we are able to identify the variations in the genes involved in the control of inflammatory process and ultimately the disease, we can successfully identify the degree of susceptibility, severity and prognosis of the disease and perhaps design medical interventions custom made according to a particular etiology. In recent past many studies have identified the association between periodontitis and variation in the genes (single nucleotide polymorphism) responsible for regulation of immune and inflammatory response.
Association of HLA (Human Leukocyte Antigens) with Periodontitis
The human leukocyte antigen (HLA) complex plays an important role in immune response. Many autoimmune diseases are found to be associated with various HLA antigens. Genes for class I and II antigens are located on chromosome number 6. At present more than 150 HLA antigens are known. The HLA molecules are involved in antigen recognition of periodontal pathogens, interaction between T and B- lymphocytes and in production of IgG. Following positive association between HLA and periodontitis was found:
- A positive association was reported between aggressive periodontitis and HLA-A9 and B15 antigens (Sofaer, 1990). Persons having these two antigens are at 3.5 times higher risk of developing the disease as compared to those who are negative for these antigens. Thus HLA -A9 and B15 seem to represent susceptibility factors for aggressive periodontitis.
- Class 2 DR4 antigen is in association with type I diabetes mellitus (Rotter et al, (1992). Since periodontitis is a diabetes related complication, DR4 antigen and periodontitis are said to be in association with each other. The DR4 antigens were found to be more prevalent in patients with aggressive periodontitis than in controls (Katz, Goultschin, Benoliel et al., 1987). However, a few studies have been unable to find any association between the two.
- HLA-A2 and HLA-B5 antigens appear to be less prevalent in patients of aggressive periodontitis as compared to controls (Kaslick, West, Chasens et al, 1975). This indicates that these HLA antigens are protective in nature.
Though above studies have indicated a positive association, many other studies have also pointed towards a negative association between HLA and periodontitis. The cause of this discrepancy appears to be the involvement of environmental factors and racial differences acting as variables.
Association between Periodontitis and Interleukin-1 (IL-1) Gene Polymorphism
Interleukin-1 plays an important role in the initiation and progression of periodontal disease. Il-1 is found in two different forms; IL-1a and IL-1b. Genes for IL-1 are present on the long arm of chromosome number 2 (2q13). IL-1 is mainly produced by activated monocytes. There is an increased level of IL-1 in periodontal tissue which stimulates bone reabsorption, inhibits collagen synthesis, up regulates matrix metalloproteinase activity and synthesis of prostaglandin.
- An initial study (Korhman et al, 1997) reported a positive association between polymorphism in the genes encoding for IL-1 and IL-1B (composite genotype) and increased severity of periodontitis. This association was seen in nonsmokers. Genotype positive nonsmokers were 6.8 times more likely to have severe periodontal disease.
- Another study (Mc Guire and Nunn, 1999) reported that the IL-1 genotype increased the risk of tooth loss by 2.7 times and heavy smoking increased it by 2.9 times. The combined effect of heavy smoking and IL-1 genotype positive increased the risk of tooth loss for 7.7 times.
- A study carried out in 2001 by Axelsson indicated that genetic polymorphism of IL-1 and smoking seems to have synergistic risk factors. These factors when combined leads not only to tooth loss but also for alveolar bone loss. Nonsmokers are at low risk of tooth loss.
(Thus there are contrasting findings regarding interactions between genetic polymorphism and smoking to affect the risk of periodontitis. Though the IL-1 genotype and smoking habit seem to interact with each other, the nature and direction of this interaction is poorly understood. More studies with large sample size are needed to evaluate the exact correlation between smoking and IL-1 interactions).
- A variant of the IL-1B coding region with a single nucleotide base pair substitution is associated with a four-fold increase in IL-1B production.
- Generalized aggressive periodontitis is in linkage disequilibrium with an allele at IL-1B. This suggests that the gene for periodontitis is located close to the gene for IL-1B (Diehl et al, 1999).
- The IL-1B allele is found to be more prevalent in patients suffering from advance chronic period-ontitis.
- Though the association between periodontitis and polymorphism of IL-1 gene cluster was observed in about 30% of European population, it was observed so only in 2% of Chinese population (Armitage, 2000).
- Many studies have indicated the lack of association between interlukin-1 polymorphism and aggressive periodontitis.
Where do we stand now? Studies in both chronic and aggressive periodontitis have yielded mixed results. It is evident that to get a clear picture of the association between IL-1 polymorphism and various types of periodontitis we need more comprehensive studies.
Tumor Necrosis Factor (TNF-α) and Periodontitis
The TNF-a is the proinflammatory cytokinase which is involved in pathogenesis of periodontitis. The TNF-α gene is located on chromosome number 6. Two different polymorphisms have been reported in the promoter region of the gene (Fassmann et al, 2003). This results in increased production of TNF-α. The level of TNF-a molecules is high at the sites of active tissue destruction and low at healthy sites. The TNF-α is the proinflammatory cytokinase
Individuals expressing the TNF-a poly-morphisms manifest greater susceptibility to certain infections. Although till date TNF-a poly-morphism has not directly been linked to susceptibility to periodontitis, there is a strong possibility that such a link exists (Craandijk et al, 2004).
Association between Neutrophil IgG Receptor (FcyR) Polymorphism and Periodontitis
FcyRs are the group of receptors that are expressed on the cell surface of leukocytes binding to IgG antibodies. The interactions between FcyRs and IgG trigger a variety of immune responses like phagocytosis, endocytosis, antibody dependant cellular cytotoxicity and enhancement of antigen presentation.
Polymorphisms in the genes coding FcyRs (especially in FcyRII) receptors (Osborne et al, 1994) are common. The FcyRII receptor is the only receptor that recognizes bacteria opsonized with IgG2. These polymorphisms result in the expression of receptors of high, low and intermediate affinities. The low affinity receptors lead to a decreased immune function and subsequently to increased susceptibility to periodontal pathogen and to severe periodontitis even in presence of high level of antibodies.
Kobayashi et al (2000) found functional poly- morphisms of IgG Fc receptors (FcyR) in Japanese patients with AgP. They found that FcyRIIIb (NA2 allele) was linked with the aggressive form of periodontitis and possibly also linked to an FcyRIIIa allele. Kobayashi et al (2001) reported that 2 FcyRIII alleles may be associated with the degree of severity in chronic periodontitis in a Japanese population.
Meisel et al (2001) analyzed the association between FeyRIIIa (high-affinity receptor) and FcyRIIIb (low- affinity receptor) and chronic periodontitis (CP). Thus a large number of studies have recognized causal association between polymorphisms of FcyR genes and both aggressive and chronic periodontitic conditions.
The alleles of FcyR gene are linked with genes causing periodontitis. The genetic poly-morphism of FeyRII receptor is a promising marker for the susceptibility of periodontitis.
Association between IgG2 Production and Periodontitis
Immunoglobulin G2 (IgG2) is produced in response to periodontal infections. The production of IgG2 is under the control of specific genes. The levels of production of IgG2 vary from person to person. The reduction in the production of IgG2 during the course of periodontal infection may lead to an increased susceptibility to disease. On the other hand an increase in the production of IgG2 provides sufficient protection against the disease. Marazita et al, (1994, 1996) studied families with aggressive periodontitis and found a major locus accounting for approximately 62% of the variance of IgG2 production.
Patients with periodontitis and normal subjects vary greatly in their capacity to produce IgG2. Patients with high titers of IgG2 antibodies have significantly less attachment loss than do patients with low titers. It is observed that patients with localized aggressive periodontitis have high titers of IgG2 compared to patients with generalized periodontitis. This indicates that IgG2 provides sufficient protection against the spread of disease and tries to limit the same.
Associations between Polymorphism in Matrix Metalloproteinase, Cathepsin C and Vit D Receptor with Periodontitis
The matrix metalloproteinase (MMP) are enzymes involved in connective tissue destruction. Single nucleotide polymorphisms are observed in MMP genes and are designated MMP-1, MMP-2, MMP-3 polymorphisms. Very few studies have evaluated the correlation between MMP polymorphisms and periodontitis (Holla et al 2005; Itagaki et al, 2004).
A recent study has indicated a decreased level of Cathepsin C activity in chronic periodontitis. Cathepsin C is a lysosomal enzyme that plays an essential role in immune and inflammatory processes and hence may play an important role in periodontitis (Hewitt et al, 2004). A mutation of the cathepsin C gene leads to Papillon-Lefore syndrome (see later).
Polymorphism in Vit D receptor has been associated with aggressive periodontitis. A very few studies, though, have verified their association.
Syndromic Form Of Periodontitis
It has been observed that there is a clear association between some type of genetically determined syndromes/disorders and periodontitis. These syndromes increase the susceptibility of the individual to periodontitis by interfering with the structural integrity of periodontal tissue or periodontitis may be a concomitant feature of the syndrome. These syndromes are due to mutations in a single gene (monogenic or Mendelian syndromes). Transmission of the disease from one generation to the next follows
Mendelian patterns of inheritance as autosomal- dominant (AD), autosomal-recessive (AR) or X-linked dominant or recessive (XLD or XLR) traits.
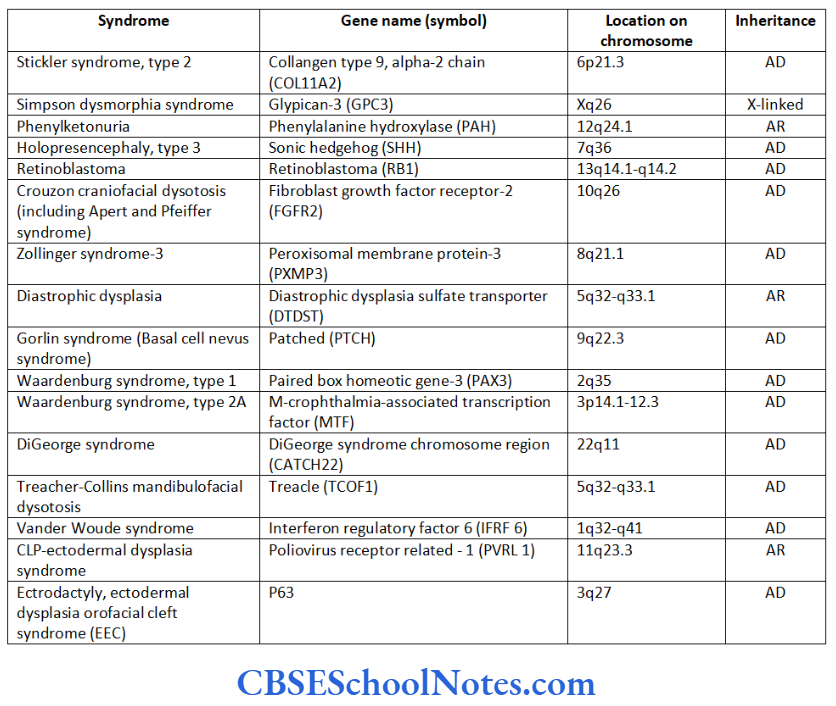
As stated above, periodontitis presents itself as a part of a wider spectrum of clinical manifestations of a number of syndromes that occur as a result of mutations in genes. This itself points towards the clearest and most direct clinical evidence that periodontitis has strong genetic etiology. Following is a brief description of some of the genetic disorders where periodontitis is observed as a clinical manifestation.
Neutrophil Functional Dispiders
Leukocyte Adhesion Deficiency Syndromes (LAD Syndromes)
Polymorphonuclear leukocytes play an important role in the restriction of the bacterial infection. There are several adhesion receptors on the surface of polymorphonuclear leukocytes. Adhesion receptors are necessary for the proper functioning of leukocytes (phagocytosis and chemotaxis). If the circulating leukocytes have defective or reduced adhesion surface receptors they will not adhere to vascular endothelium.
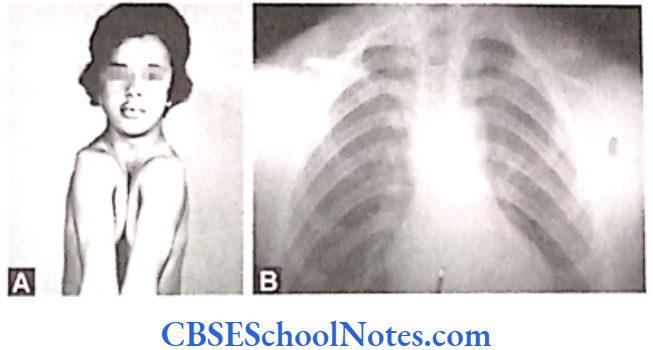
As a result leukocytes would not be accumulated at the site of inflammation where they are needed to combat the infection. This leads to an increased susceptibility to an infectious disease as exemplified by the leukocyte adhesion deficiency syndrome (LADS). Recurrent bacterial infections, impaired pus formation and impaired wound healing clinically characterize the syndrome. A number of these infections are associated with increased susceptibility to periodontitis. LAD occurs in two forms, LAD syndrome type I and LAD syndrome type 2.
Inheritance: Both types of LAD syndromes are inherited as AR.
Mutation in LAD type 1: The mutation occurs in the B2 integrin chain (ITGB2) gene -21q22.3 (Arnaout et al, 1990). At present more than 20 mutations of the integrin B2 gene are known. The gene mutation causes defects in the integrin receptors of leukocytes. This mutation leads to defects in cell adhesion and chemo- taxis ultimately resulting in increased susceptibility to severe infections including prepubertal aggressive periodontitis.
Mutation in LAD type 2: This is a rare variety of the LAD syndrome and is usually associated with psychomotor retardation (mental retardation, short height and recurrent infection). The syndrome is caused due to a mutation in the guanosine 5′- diphosphate-fucose transporter 1 (SLC 35C1) gene (Lowe et al, 1990; Ishikawa et al, 2005). The infectious episodes and severity of disease is much milder as compared to LAD type I. This syndrome results in chronic periodontitis.
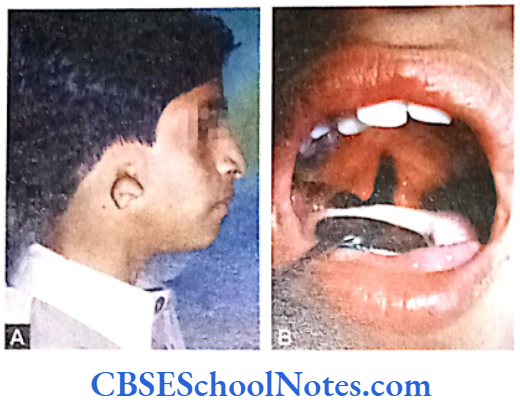
Chédiak-Higashi Syndrome
The syndrome is clinically characterized by a decreased pigmentation of eyes and hair, photophobia, nystagmus and susceptibility to infection.
Inheritance: This syndrome is inherited as an autosomal recessive (AR) trait.
Mutation: The mutation occurs in lysosomal trafficking regulator (LYST) gene-1q42.1-q42.2 (Nagle et al, 1996). This leads to an abnormal transport of vesicles to and from neutrophil lysosomes or to a defect in the ability of cells to produce lysosomes (Charette et al, 2007). Chédiak-Higashi syndrome is associated with severe periodontitis unresponsive to conventional periodontal treatment. However, periodontitis is only seen in its severe form with the syndrome.
Deficiency in Neutrophil Number (Neutropenias)
Neutropenia is defined as an abnormally low number of circulating neutrophils. The condition is associated with increased susceptibility to infections such as aggressive periodontitis. Table 14.1- includes various types of neutropenias and associated gene mutations in brief.
Genetic Defects of Structural Components
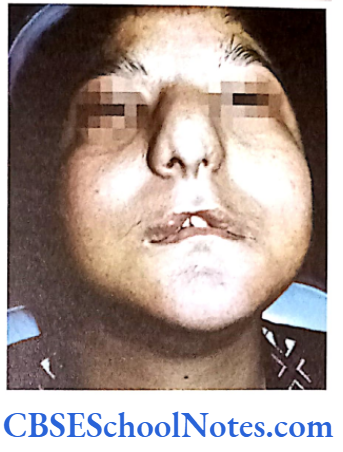
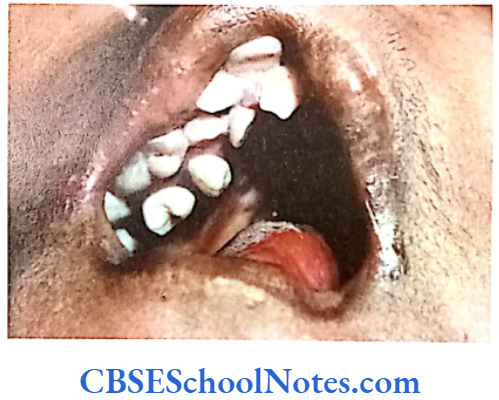
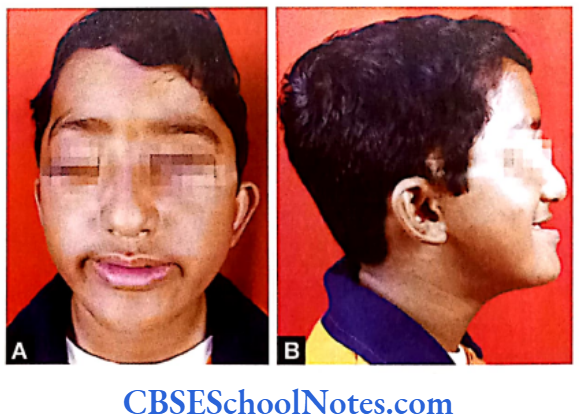
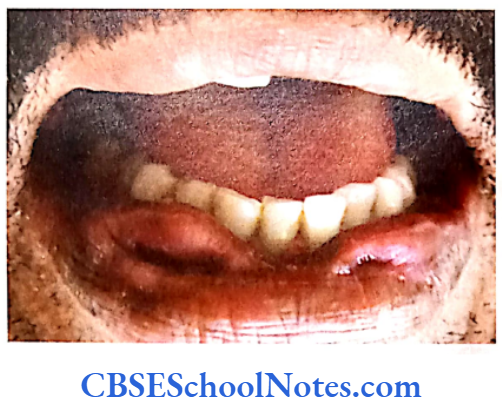
Papillion-Lefèvre Syndrome (PLS)
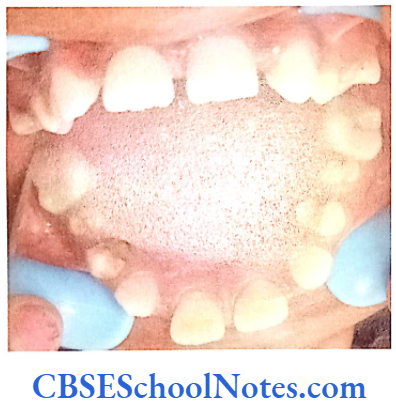
This syndrome is characterized by severe early onset periodontitis, which affects both primary and permanent dentitions and hyperkeratosis of palmar and plantar surfaces.
Inheritance: Autosomal recessive inheritance (AR).
Mutation: It is due to mutations in the cathepsin C gene which is located on chromosome 11 (11q14-q21). At present more than 50 mutations have been identified in cathepsin C gene. Cathepsin C is a cysteine protease that plays a role in degrading proteins and activating proenzymes in immune and inflammatory cells. Inactivation of cathepsin C results in failure to cleave and activate the neutrophil serine proteases cathepsin G, neutrophil elastase and proteinase 3 (Pham et al, 2004).
It was observed that in some of the PLS patients aggressive periodontitis were associated with a kind of virulent microorganism. Elimination of this microorganism prevented the periodontal destruction. This suggested that the periodontitis was not a direct effect of mutation of the gene but the gene mutation increased the susceptibility of a person to infection by the specific microorganism instead.
Ehlers-Danlos Syndrome (EDS)
This syndrome is associated with connective tissue disorders. EDS is characterized by defective formation of collagen fibers. The abnormal collagen in the patient leads to fragility and hyperextensibility of the skin, hypermobility of joints, easy bruising and is also associated with early onset of periodontitis. Though at least 17 different types of EDS have been reported but early onset periodontitis has been associated only with two of its subtypes: EDS type 4 and EDS type 8.
Inheritance: Both the types 4 and type 8 EDS are inherited as autosomal-dominant (AD) conditions.
Mutation: The mutation leading to the type 4 EDS was found to be linked to the gene responsible for the synthesis of type 3 (COL 3A1) collagen (Superti-Furga et al, 1988). This leads to the defect in synthesis of collagen 3 resulting in decreased levels of collagen 3.
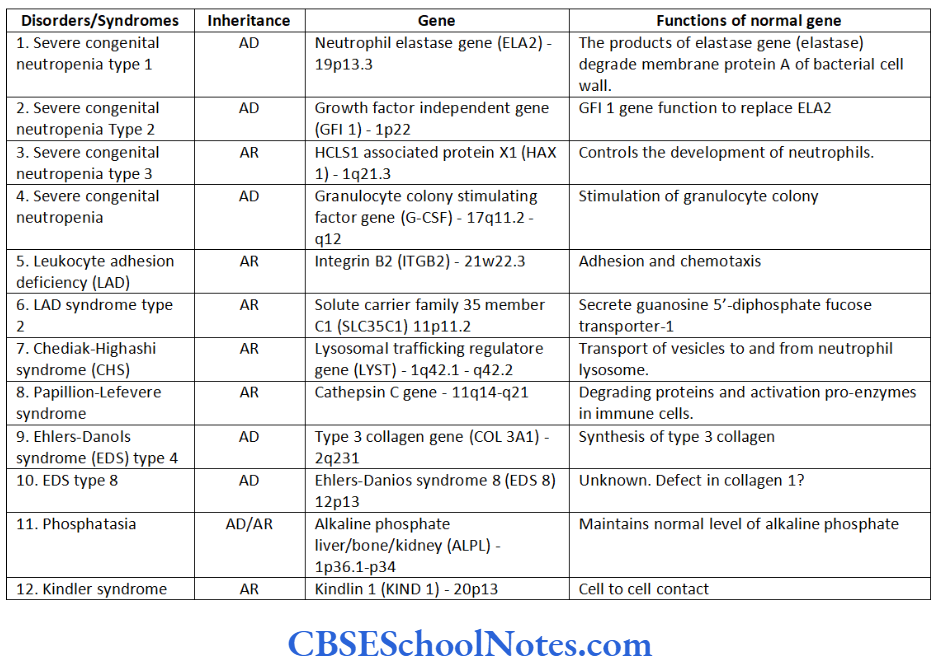
Mutations causing type 8 EDS, though, have not yet been identified. This disease has been, nevertheless, linked to chromosome number 12 (12p13). The association of early onset periodontitis with type 8 EDS is more common as compared to that of type 3.
Hypophosphatasia
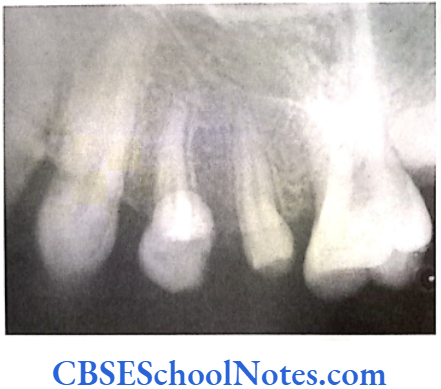
These patients have decreased levels of alkaline phosphatase in the serum. The condition is associated with abnormal bone mineralization, skeletal abnormalities and cementum hypoplasia. These patients present a severe loss of alveolar bone and premature loss of primary teeth. Pulp chambers may also get enlarged. Lack of connective tissue attachment to bone is responsible for early exfoliation of the primary teeth. Hypophosphatasia may be considered as an etiology of aggressive periodontitis. The condition leads to premature loss of primary teeth and occasionally permanent teeth.
(Mode of) inheritance: Autosomal dominant or recessive.
Mutation: Mutation occurs in the ALPL (alkaline phosphatase, liver/kidney/bone) gene. The gene is localized on the short arm of chromosome 1 (1p36.1- p34). Mutations result in abnormal production of alkaline phosphatase leading to poor mineralization of tissues.
Kindler Syndrome
It is caused due to a mutation in the Kinderlin gene (KIND1) (Jobard et al, 2003). The gene is expressed in many tissues including epidermal keratinocytes. In skin it plays a role in cell adhesion process. Patients of Kindler syndrome show multiple dermatological findings (hyperkeratosis, pigmentation, eczema, skin fragility, etc.), the syndrome is associated with severe aggressive periodontitis both in the primary and secondary dentition.
(Mode of) inheritance: Autosomal recessive (AR) in nature.
Mutation: Mutation occurs in the Kindlin gene (KIND1) situated on the short arm of chromosome number 20 (20p13).
From the above description one can conclude that isolated periodontitits is clearly behaves as a multifactorial disease. Researchers have shown associations between complex form of periodontitis and abnormalities in genes (single nucleotide polymorphism) responsible for regulation of immune and inflammatory responses. Further research is needed to examine the role of both environmental and genetic factors in the causation of periodontitis.
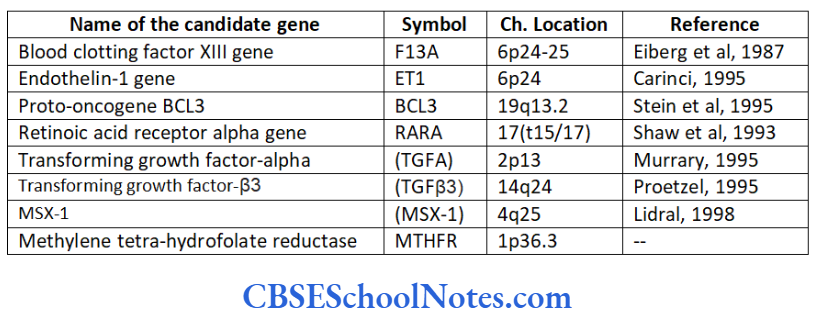
We have also seen that monogenic (single gene) inheritance of periodontitis is possible but these incidences are associated with certain syndromes. We have successfully located many mutant genes (refer table) and understood to some extent the mechanisms of these mutations that disrupt normal homeostasis in the periodontum.
Though a large number of studies are being conducted to identify the genetic basis of periodontitis, we are still far away from determining the risk factors, prevention and treatment of aggressive and chronic periodontitis, genetically.