
Types Of Impurities In Pharmaceutical Substances Introduction
Pharmaceutical Chemistry is the branch of chemistry which deals with the study of medicines. It includes the main branches of chemistry namely inorganic chemistry, organic chemistry, medicinal chemistry, analytical chemistry and physical chemistry.
The term “Inorganic Chemistry” in shorter sense is called “chemistry of everything else”. It deals with the study of inorganic compounds containing their molecular weight, molecular formula, occurrence, method of preparation, physical and chemical properties, assay and uses as pharmaceutical aids, therapeutic and diagnostic agents.
It has accomplished rapid progress in understanding the properties of all chemical compounds. It is of fundamental importance not only as a basic science but also as one of the most useful source of modem technologies.
The pharmaceutical substances used in the treatment of diseases may be procured from the natural sources such as minerals, salts, purified inorganic chemicals, calcined inorganics (ash or bhasma), plants, animals, microbes and from synthetic chemicals.
In ancient times mercury, gold, copper and silver were also used in medicine. The specification for each pharmaceutical is listed in Pharmacopoeia.
Pharmacopoeia
The term Pharmacopoeia is derived from the Greek word “pharmakon” means drug or medicine and “poiein” is to make. Drugs manufactured in India have to be labelled with the mandatory non-proprietary name with the suffix “I.P”. It provides a collected list of drugs and medicinal substances along with directions for making preparation from them.
Many countries are now publishing pharmacopoeias of their own and by this they are able to control the standards of the drug produced in their countries and also the standards of the drugs imported into their countries and ensure the health of their people.
A drug or a medicinal chemical included in a pharmacopoeia is termed as official and the sections dealing with official drugs, preparation and substances are known as monograph.
Read and Learn More Pharmaceutical Inorganic Chemistry Notes
History of pharmacopoeia
Indian Pharmacopoeia (I.P.)
In India, the first pharmacopoeia had been publishes as “Bengal Pharmacopoeia in 1844.
The Government of India constituted a permanent Indian Pharmacopoeia Committee in 1948 for preparing Indian Pharmacopoeia under the chairmanship of Dr. B. N. Ghosh. The first edition of I.P. was publishes in 1955, followed by a supplement in 1960 which contains large number of crude drugs and their preparation.
List of Publication of various Edition of Indian Pharmacopoeia

Salient features of First edition of Indian Pharmacopoeia (1955):
- Covers 986 monographs.
- Titles of monograph in Latin language.
- Weight and measures in metric system.
- Doses expressed in both metric and English system.
- List of preparations given at the end of some of the monographs.
- Abbreviated titles used.
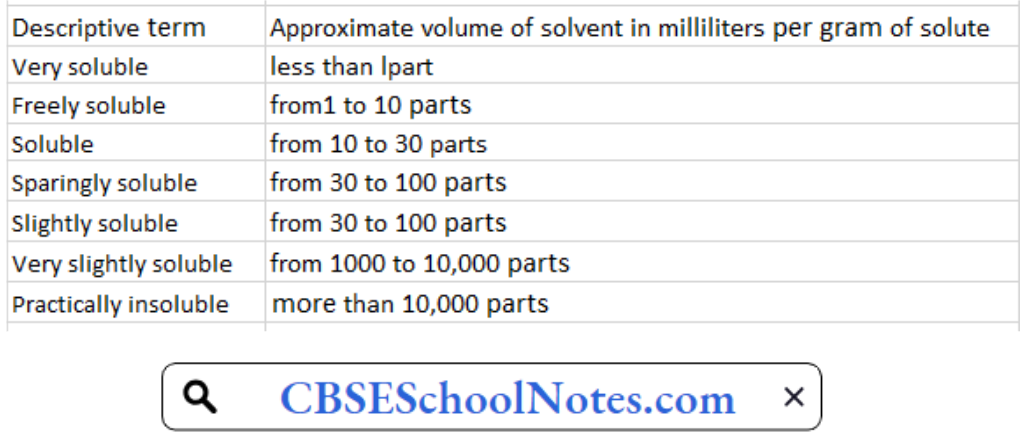
- Descriptive terms used for solubility instead of exact solubility.
Salient features of Second edition of Indian Pharmacopoeia (1966):
- Published in 1966 and its supplement was published in 1975.
- Titles of monographs changed from Latin to English.
- Name of drug comes first in title e.g. Aminophylline Injection.
- Solubility expressed in parts of solvent per unit part of solute.
- 93 new monographs were added.
- New analytical techniques had been included.
- Test for sterility had been modified to detect fungi.
- ‘Usual Strength’-a heading used to represent strength of tablet had been used.
- 214 monographs from I.P. 55 were deleted.
Salient features of Third edition of Indian Pharmacopoeia (1985):
- Published in 1985 with two volumes and nine appendices.
- 260 new monographs were added and 450 were amended.
- New analytical techniques (Flame photometry,
- Electrophoresis, Fluorometry etc.) had been introduced.
- Dissolution had been introduced.
- Microbial limit tests being prescribed for liquid preparation.
- Gas liquid chromatography had been recognized.
- Determination of viscosity have been modified involving use of Ostwald viscometer.
- New appendix “Water for Pharmaceutical Use” had been introduced.
- Drugs renamed and some drugs had been omitted.
- Addendum I to I.P. 1985 was published in 1989, where 46 new monographs were added and 126 amended.
- Addendum II to I.P. 1985 was published in 1991, where 62 new monographs were added and 110 amended.
Salient features of Fourth edition of Indian Pharmacopoeia (1996):
- Published in 1996 under the chairmanship of Dr. Nityan and along with Dr. Parvinder Singh followed by its addendums in 2000 and 2003.
- Contains 1149 monographs and 123 appendices in two volumes.
- Computer generated structural formulae used.
- Infrared and Ultra Violet absorption spectrophotometric tests for identification of drug were added.
- Included 294 new monographs and 110 monographs were deleted.
- High Pressure Liquid Chromatography (HPLC) had been used as analytical method.
- Bacterial endotoxin test were introduced.
- Quantitative method for determining particulate matter in injectable preparation being replaced by qualitative.
- Monographs of ORS, ORS-bicarbonate had been omitted out and ORS-citrate formula had been added.
- Specific biological assay and test were transferred to individual monographs.
- The veterinary supplement of I.P. 1996 contains 208 monographs and four appendices.
Salient features of Fifth edition of Indian Pharmacopoeia (2007):
- Published in 2007 followed by its addendum in 2008.
- Presented in three volumes.
- Volume one contains general notices, structure of IPC, Acknowledgements.
- Volume two and three contains general monographs on dosage forms, drug substances and pharmaceutical aid.
Salient features of Sixth edition of Indian Pharmacopoeia (2010):
- Published in 2010 followed by its addendum in 2012 and DVD of I.P. 2010 was also available.
- Released on 27th December 2011 by Mr. P.K. Pradhan in presence of Mr. L. C. Goyal and Dr. Arun Kumar.
- Consists of three volumes.
- Volume one contains Notices, preface, structure of IPC, general chapters.
- Volume two contains general monographs on dosage form, drug and pharmaceutical aid (A to M).
- Volume three contains general monographs on dosage form, drug and pharmaceutical aid (N to Z).
- Products of biotechnology, herbal products, and additional antiretroviral drugs were included.
- Standards for new drug under NHP were added.
- Microbial contamination chapter updated.
- Chapter on NMR is incorporated in appendices.
- New chapter on Liposomal products is also added.
Salient features of Seventh edition of Indian Pharmacopoeia (2014):
- Published in 2014 and presented in four volumes.
- Contain 2567 monographs of drugs out of which 577 are new monographs.
- Introduced 19 radiopharmaceutical monographs for the first time.
- 10 antibiotic monographs, 31 herbal monographs, 5 vaccine, and immunosera for human use, 6 insulin products and 7 biotechnological products with 19 new general chapters were included.
Salient features of Eighth edition of Indian Pharmacopoeia (2017):
The latest Edition of Indian Pharmacopoeia, IP-2018 has been released on 29th September, 2017 by Sh. C.K. Mishra, Secretary Health & Family Welfare, Govt, of India.
IP-2018 has been brought out in 4 Volumes incorporating 220 new monographs (Chemical Monographs (170), Herbal Monographs (15), Blood and Blood related products (10), Vaccines and Immunosera for Human use monographs (02), Radiopharmaceutical monographs (03), Biotechnology Derived Therapeutic Products (06), Veterinary monographs
(14)), 366 revised monographs and 7 omissions.
Salient Features of IP-2018:-
- General Chemical tests & Thin Layer Chromatography (TLC) for identification of an article have been almost eliminated and more specific infrared, ultraviolet
spectrophotometer and HPLC tests have been given emphasis. The concept of relying on published infrared spectra as a basis for identification has been continued. - The use of chromatographic methods has been greatly extended to cope with the need for more specificity in assays and in particular, in assessing the nature and
extent of impurities in ingredients and products. - Most of the existing Assays and Related Substances Test methods are upgraded by liquid chromatographic in view to harmonize with other International Pharmacopoeia.
- Pyrogen test have been replaced by Bacterial Endotoxin test (BET) in parenteral preparations and other monographs.
- For ease of access to make Pharmacopoeia more user friendly, Index has been incorporated in Volume-I along with that already existing in Volume-IV of IP.
- 53 New Fixed Dose Combination (FDC’s) combination monographs have been included, out of which 25 FDC monographs are not available in any Pharmacopoeia.
- General Chapters on Volumetric Glassware, Conductivity, Dissolution test, Disintegration test, Dimensions of Hard Gelatin Capsule Shells etc. have been revised.
- For Controlling the Microbial quality of all the medicinal product general chapter on Maintenance, Identification, Preservation and Disposal of Microorganism have been
revised.
British Pharmacopoeia (B. P.)
British Pharmacopoeia was published by the health ministers of the United Kingdom. The first edition of B.P. was published in 1864 which consists of two parts “Materia Medica” and
“Preparation and Compound”. Australia and Canada are two of the countries that have adopted the B.P. as their national standard book. The various publication of B.P. comes out in subsequent years as under:
- 1867: Second edition.
- 1885: Third edition.
- 1898: Fourth edition.
- 1914: Fifth edition.
- 1932: Sixth edition.
- 1948: Seventh edition. It contains official 49 tablet preparations and official 75 injections.
- 1951: Addendum to BP 1948 was published.
- 1953: Eighth edition. In this title of drug and preparation was in English instead of Latin and metric system was used.
- 1955: Addendum to BP 1953 was published.
- 1958: Ninth edition.
- 1960: Addendum to BP 1958 was published. It includes monographs on radioactive chemicals.
- 1963: Tenth edition.
- 1968: Eleventh edition.
- 1973: Twelfth edition.
- 1980: Thirteenth edition.
- 1988: Fourteenth edition.
- 1988, 1990, 1991 & 1992: Four addendums were published.
- 1993: Fifteenth edition.
- 2000: Sixteenth edition. It contains veterinary drugs.
The other editions were published in 2004, 2005, 2007-2009 (published in six volumes), 2010 and 2014. The current edition was published in 2015 which includes almost 3500 monographs spread out in six printed volumes.
United State Pharmacopeia (U.S.P.)
United States Pharmacopeia and the National Formulary (USP-NF) are recognized as official compendia for determining the standard of pharmaceutical products. The first USP was published in 1880 under the authority of United State Pharmaceutical Convention (USPC).
National Formulary (NF) was published first in 1888 under the authority of the American Pharmacists Association (APhP). After 1975, both USP and NF are published by USPC in a
combined volume as USP-NF.
USP-NF was published at every ten years interval from 1820-1942. But from 1942-2000, it was published at an interval of five years. After 2000, USP-NF
has been published annually. USP 40 – NF 35 will be officially released in year 2017.
British Pharmaceutical Codex (B.P.C.)
This book was prepared as a reference book for the use of medical practitioners and dispensing pharmacists. In 1903, Pharmaceutical society of Great Britain came up with BPC. The first edition of the BPC was published in 1907.
The subsequent editions were published in 1911, 1923, 1934, 1949, 1954, 1959, 1963, 1968 and 1973. From eleventh edition 1979 onwards BPC was considered as Pharmaceutical Codex only. It comprises of general monographs of drugs and also provides standards of medicaments and materials that not included in BP and British National Formulary (BNF).
Extra Pharmacopoeia (MARTINDALE)
This pharmacopoeia was first issued in 1883 by William Martindale and now it was published by pharmaceutical society of Great Britain.
Pharmacopoeial description
Practically most of the pharmacopoeia consists of three main sections namely:
- General notices
- Monographs of the official drugs
- Appendices
General notices: It is useful information of pharmaceutical progress since last edition as it summarizes the various changes including additions/deletions in the present edition
compared to last edition.
Monographs of the official drugs: The word “Monograph” means the written study of a subject. It is derived from a Greek word (mono=single, grapho = to write). As the medicinal substances are to be used for the cure and prevention of diseases, therefore,these are considered as very important and hence their written studies appear as monograph .
- Titles:- Monograph titles are in English and French in the respective versions and there is a Latin subtitle. It includes the main names of the substance.
- Synonyms: Common names, if any, of the substance.
- Chemical formula and Molecular Weight: If necessary graphic, molecular formula and molecular weight are given at the beginning of the monograph.
- Chemical names:- It is given in the monograph as employed by the International Union of Pure and Applied Chemistry (IUPAC). It does not constitute for the analytical standards such as UV, NMR, IR etc.
- Category: It indicates its use in pharmacy/medicine. It represents pharmacological action of the substance such as antifungal, antimicrobial, anti inflammatory, etc.
- Doses: Doses mentioned in the Indian Pharmacopoeia (I.P.) represent the average range of quantities suitable for adults.
- Description: This include a statement about its general physical properties , i.e. whether the substance is a liquid or solid, coloured or colourless, amorphous or crystalline, its taste, etc.
- Solubility: In terms of solubility the terms used have the following significance referred to a temperature between 15ÿC and 25ÿC. The term “partly soluble” is used to describe a mixture where only some of the components dissolve.
The term “miscible” is used to describe a liquid that is miscible in all proportions with the stated solvent. - Standards: Indian Pharmacopoeia (I.P.) prescribes the standard of purity and strength in the monograph of almost all official substances. A substance is not deemed to be of
standard quality unless it complies with the requirements stated under ‘standard’ of its monograph. - Identification: It includes some specific and non-specific test of the substance.
- Test for purity: Indian Pharmacopoeia (I.P.) prescribes tests for purity of almost all the offcial substances. These tests include boiling point, melting point, limit test for chlorides, sulphates, iron, aresenic, lead, heavy metals, pH of solution, loss on drying, specific optical rotation, etc.
- Assay: Assay is used for the quantitative determination of principal ingredients of the official substances and their preparations.
- Storage:-Indian Pharmacopoeia prescribes the conditions for storage in such a way as to prevent contamination and, as far as possible, deterioration. Where special conditions of storage are recommended, including the type of container and limits of temperature, they are stated in the monograph. The following expressions are used in monographs under Storage with the meaning shown.
- In an airtight contaitter means that the product is stored in an airtight container. Care is to be taken when the container is opened in a damp atmosphere. A low moisture content may be maintained, if necessary, by the use of a desiccant in the container provided that direct contact with the product is avoided.
- Protected from light means that the product is stored either in a container made of a material that absorbs light sufficiently to protect the contents from change induced by such light or in a container enclosed in an outer cover that provides such protection or stored in a place from which all such light is excluded.
- Labelling When the term “label” is used in the Pharmacopoeia, the labelling statements may appear on the container, the package, a leaflet accompanying the package or a certificate of analysis accompanying the article, as decided by the competent authority.
Appendices: General notices and monographs are followed by comprehensive section of appendices.
Appendix- I: It describes about the apparatus needed for various pharmacopoeial tests and assays.
Appendix-ll: It describes about biological tests and assays.
Appendix-Ill: It contains details of various chemical tests and assays.
Appendix-lV: It contains details of microbiological tests and assays.
Appendix-V: It includes physical tests and determinations like loss on drying, pH determination, melting range, etc.
Appendix-Vl: It describes useful directions on cleaning glassware’s.
Appendix-VIl: It describes the reagents and solutions needed for various tests and assays.
Appendix-VIII: It describes about the reference substances.
Appendix-IX: It describes the names and symbols used in the pharmacopoeia and their atomic weights have been described.
Impurity
Impurity is defined as any substance coexisting with the original drug, such as starting material or intermediates or that is formed, due to any side reactions. Chemical purity means
freedom from all foreign materials. Purification of chemicals is expensive and therefore purifying a substance to much higher degree is necessary.
Effects of Impurities
Pure substances are difficult to get and some amount of impurity is always present in the material. So the impurities which are present in the substances may have the following effects:
- Impurities may lower the shelf life of the substances.
- Therapeutic effect can be decreased.
- Impurities may bring about incompatibility with other substances.
- Impurities may cause1difficulties during formulations and use of the substances.
- Sometimes Impurities changes the chemical and physical properties of the substances.
- Shows toxic effect after a certain period.
Sources of Impurity:
A compound having foreign materials is said to be impure. The origin of impurities in drugs is from various sources and phases of the synthetic process and preparation of pharmaceutical dosage forms. Majority of the impurities are characteristics of the synthetic route of the manufacturing process. The pharmaceutical preparation should be free from toxic and other impurities.
The impurities commonly found in medicinal preparations are:
- Impurities due to which substances become incompatible.
- Due to colouring or flavouring substances, e.g., Sodium Salicylate.
- Humidity.
- Chemical and physical properties.
The various sources of impurities in pharmaceutical substances are as follows:
Raw Materials Used in the Manufacturing of Pharmaceutical Process: The source of pharmaceutical substances are either natural or synthesized from chemical starting materials. It is essential to verify the identity of the source material and its quality otherwise it contaminate the final product. Example: lead and heavy metals are found as impurities in many sulphide ores, Rock salt used for the preparation of sodium chloride is contaminated with small amounts of calcium and magnesium chlorides.
Reagents employed in the manufacturing process: Pharmaceutical substances are either isolated from natural sources (mineral sources, plants, animals and microbes) or synthesized from chemical starting materials. If reagents are employed in the manufacturing process are not completely removed by washing, these reagents may be present in final products.
Example: e.g., Magnesium impurities are found in calcium minerals, aluminum ores are usually accompanied by alkali and alkaline earth compounds.
Solvents: Water is the most commonly used solvents in the preparation of inorganic pharmaceuticals. Different types of water containing different types of impurities. Various types of water are:
Tap water: Containing impurities of Magnesium, sodium, calcium, chloride, sulphates and carbonates. +
Softened water: It is prepared from tap water nut it contains more of sodium and chloride ions as impurities.
Demineralized water: It is prepared by ion exchange and it is free from Magnesium,sodium, calcium, chloride, sulphates and carbonates impurities. It contains pyrogens, bacteria and organic impurities.
Distilled water: This water is free from all inorganic and organic impurities. It is best solvent for pharmaceutical preparations.
Action of reagents on reaction vessels: Reaction vessels used in the manufacturing process may be metallic such as iron, cast iron, galvanized iron, copper, silver, aluminium, nickel, zinc and lead.
Atmospheric contamination during manufacturing process: Atmosphere may contain dust (sulphur, aluminum oxide, silica, soot etc.) and some gases like carbon dioxide, sulphur dioxide, arsine and hydrogen sulphide. These may contaminate the final product during the manufacturing process.
Chemical process used in the manufacture: Various chemical reactions such as oxidation, nitration, reduction, halogenations, hydrolysis are involved for the synthesis of drugs. In these chemical reactions various chemicals and tap water is used it is often having Mg+2, Ca+2 and Ch, which are generally found in the substance which is being manufactured.
Defects in the manufacturing process : Defects like incompleteness, pH, pressure, temperature and imperfect mixing in various manufacturing processes produce impurities in chemical compounds.
To prevent these impurities many test such as limit test are carried out to lower the impurities and to make the pharmaceuticals more safe.
Limit Test
Limit test is defined as quantitative or semiquantitative test designed to identify and control small quantity of impurity which is likely to be present in the substance.
Limit test is generally carried out to determine the inorganic impurities present in the compound.
Limit test for chlorides, sulphates, iron, lead and heavy metal are carried out in Nessler cylinders. Nesseler cylinders are made up of borosilicate glass that is colourless. It has the fixed diameter, length as per according to the Indian Pharmacopeia.
Two similar kind of cylinders are required each time i.e. one for the ‘Test’ sample and other for the ‘standard’ to make comparison in the identical manner. The quantities of the sample vary according to the , limits of impurities while the standard remains constant.
No numerical values for ths limits in these tests are prescribed in the pharmacopoeias, as it is not practicable. Generally an aqueous solution of the substance is prepared.
Sometimes a solution of the, substance is prepared by dissolving in an acid or if the solution is alkaline it is neutralized with nitric or hydrochloric acid as specific in the monograph of the pharmacopoeia. The Extent of opalescence, turbidity and colour is affected by other impurities present in the substance.
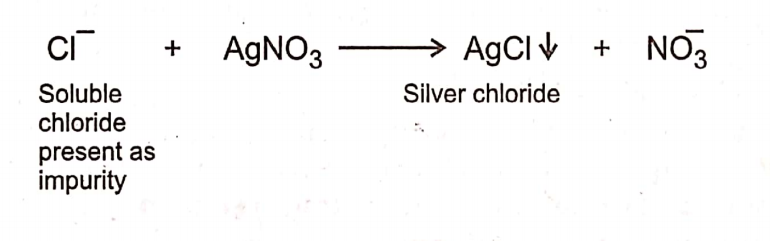
Limit Test for Chloride
Limit test of chloride is based upon the chemical reaction between silver nitrate and soluble chlorides in presence of dilute nitric acid to give opalescence (solid particles) of silver
chloride. Opalescence produced is compared with the standard solution. If the opalescence in the sample is less than the standard, it passes the test. If it is more than the standard, it fails the test.
Procedure
Take two 50 ml Nessler Cylinders.-Label one as “Test” and- the other as ‘Standard’.

Then observe both Nesseler cylinder from the side and from above against a black background and compare the turbidity. The turbidity developed in the test solution is not thicker than that of standard solution.
Limit Test for Iron
Limit test of iron is based on the reaction of iron with thioglycolic acid in the presence of citric acid and ammonia when pale pink to deep reddish purple colour is produced. Citric acid forms a complex with iron and prevent its precipitation by ammonia as ferric hydroxide. It will result to the formation of purple coloured ferrous salt of thioglycolic acid.
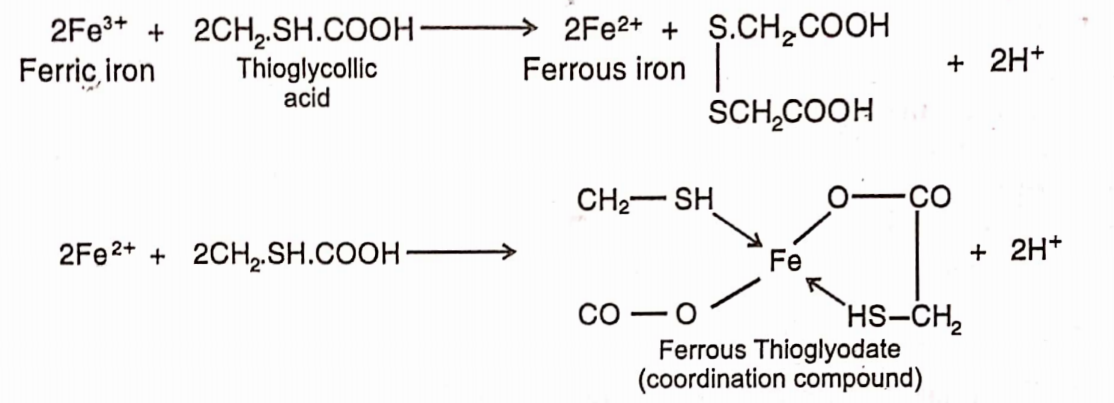
The colour produced is due to the formation of a ferrous compound with thioglycolic acid. It is stable in the presence of air but fades when exposed to air due to oxidation to the
ferric compound.
Standard Iron Solution: Accurately 0.1726g of ferric ammonium sulphate is weighed and dissolved in 10ml of 0.1N sulphuric acid and sufficient water to produce 100ml. Each ml of this solution contains 0.02mg of iron.
Procedure
Take two 50ml Nesseler cylinders. Label one as the ‘Test’ and the other as ‘Standard’.
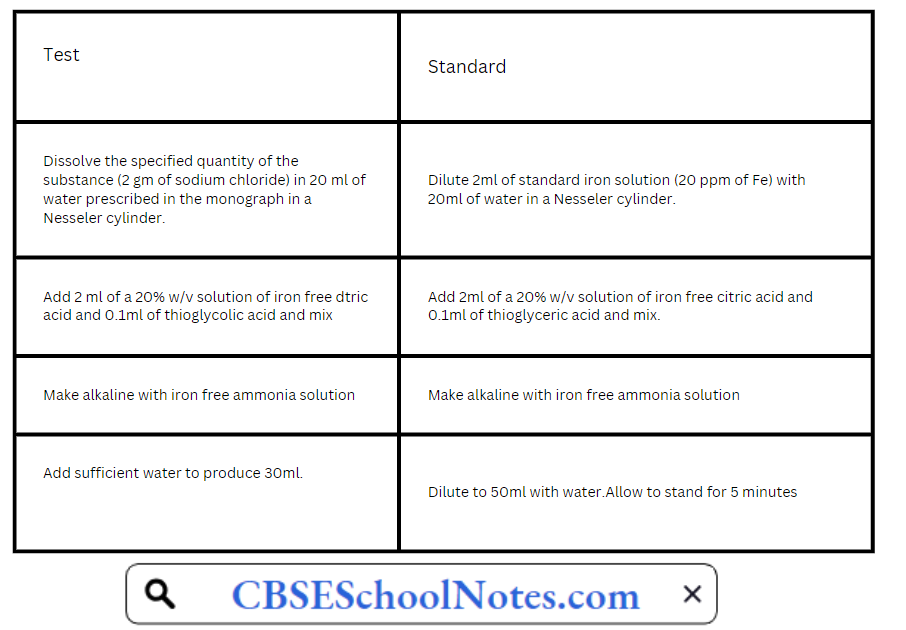
Compare the purple colour in two Nesseler cylinders by viewing vertically downwards. Any colour produced in test solution should not be more intense than the standard solution.
If the intensity of the colour is more in test than in the standard, it means that the sample contains more quantity of iron impurity than the permissible limit and hence the sample is
declared as not of standard quality
The colour in the test and the standard should be compared immediately after five minutes allowed for full development of the colour is over. If there is any delay, the colour
fades due to oxidation and the test becomes unreliable.
List of substances for which limit test of iron is prescribed in I.P. 1996.
- Calcium carbonate
- Calcium chloride
- Zinc oxide
- Zinc sulphate
- Heavy magnesium carbonate
- Light magnesium carbonate
- Sodium acetate
- Sodium bicarbonate
- Sodium chloride
Limit Test for Sulphate
Limit test of sulphate is based on the reaction of soluble sulphate with barium chloride in presence of dilute hydrochloric acid to form barium sulphate which appears as solid particles (turbidity) in the solution.
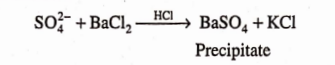
Hydrochloric add is added to prevent precipitation of other acid radicals such as phosphate, oxalate etc. by common ion effect with barium chloride sol. So that less barium ions are formed.
Reagents:
Barium Sulphate Reagent: 15ml of a 0.5M barium chloride, 55ml of water and 20ml of sulphate-free alcohol are mixed, 5ml of a 0.0181% w/v solution of potassium sulphate are added, diluted to 100ml with water and mixed. Barium sulphate reagent must be freshly prepared.
0.5M Barium Chloride: Barium chloride dissolved in water to contain in 1000ml 122.1g ofBaClÿHjO.
Procedure: Take two 50ml Nesseler cylinders. Label one as “Test” and the other as “Standard”.
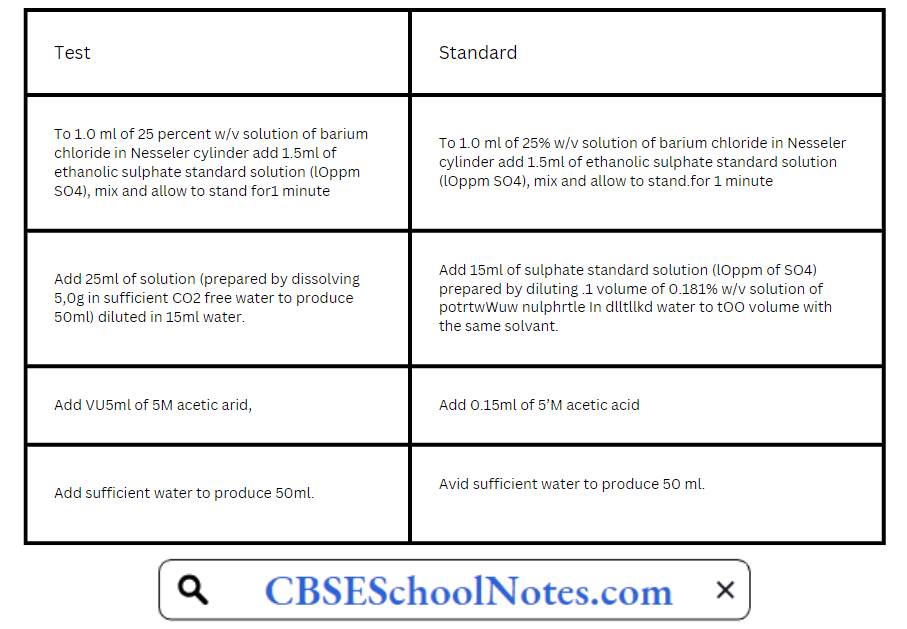
Stirr immediately each solution with a glass rod and allow to stand for 5 minutes, Compare the turbidity transversally against a black background in two Nesscler cylindres. Any colour
produced in test solution should not he more intense than the standard solution.
LimitTest for Heavy Metals
The limit test for heavy metals is based on the reaction between the solution of heavy metals and a saturated solution of Hydrogen sulphides. In acidic media, it produces reddish/black colour with hydrogen sulphide which is compared with standard lead nitrate solution.

It is designed to determine tire content of metallic impurities that are coloured by sulphide ion, under specified conditions. It is indicated in the individual monographs in terms of lead per million parts of tire substance (by weight), as determined by visual comparison of tire colour produced by the substance with that of a control prepared from a standard lead solution.
The amount of heavy metals is determined by one of tire following methods and as directed in the individual monographs. Method A is used for substances that yield clear, colourless
solutions under the specified test condition.
Method B is used for substances that do not yield dear, colourless solutions under tire test conditions specified for method A, or for substances which, by virtue of their complex nature, interfere with the precipitation of metals by sulphide ion. Method C is used for substances that yield clear, colourless solutions with sodium hydroxide solution.
Method A:

The colour produced in the test solution should not be darker than that produced in the standard solution.
Method B:
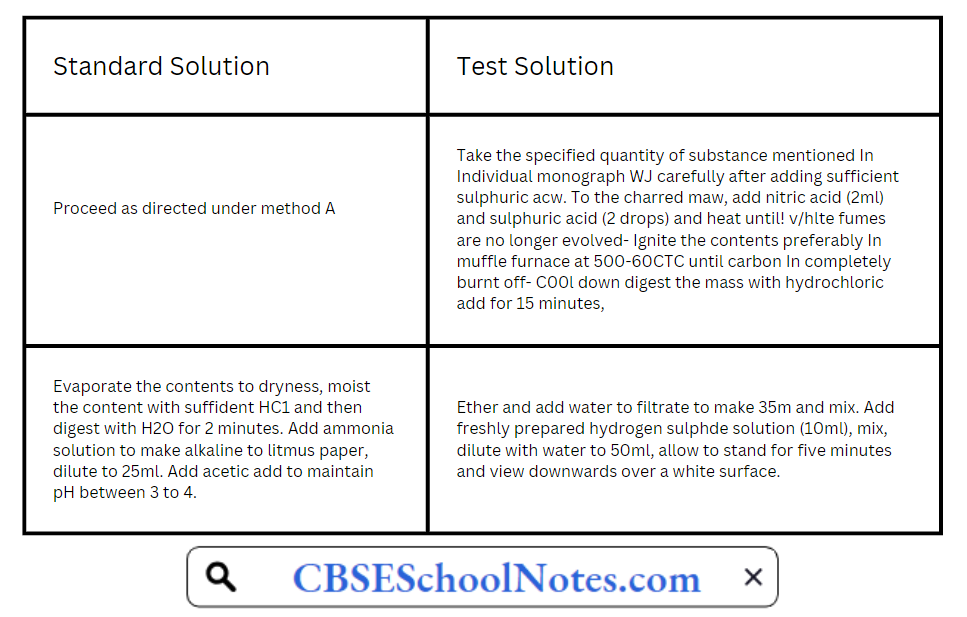
Method C:
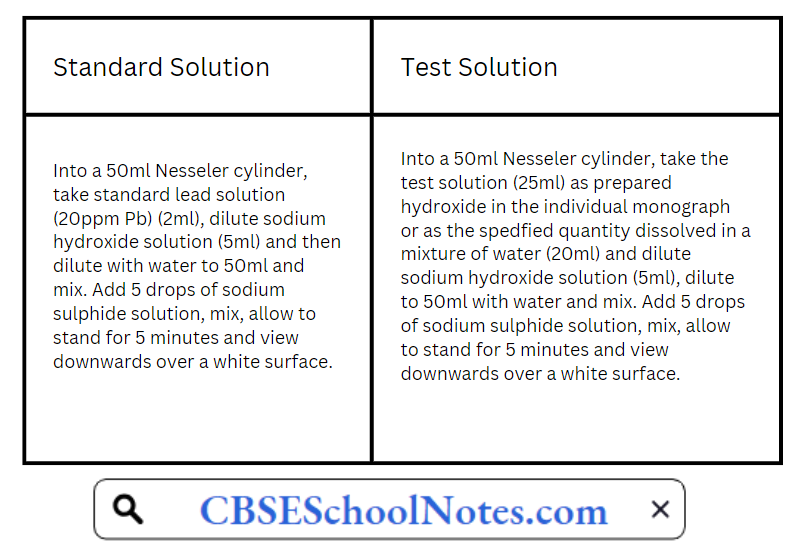
The colour produced in the test solution should not be darker than the colour of the standard solution. Lead standard solution (20ppm Pb) is prepared by diluting 1 volume of lead standard solution (100 ppm Pb) to 5 volume with water.
Lead standard solution (100 ppm) itself is prepared by diluting 1 volume of lead standard solution (0.1% Pb) to 10 volume with water Lead standard solution (0.1% Pb) is prepared by dissolving 0.400 gm of lead nitrate in water containing 2 ml of cone. Nitric acid & adding sufficient water to produce 250 ml.
Limit Test for Lead
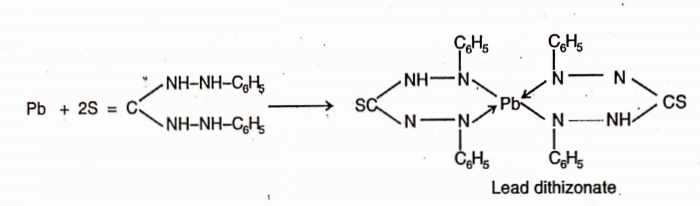
Lead is a toxic substance present in pharmaceutical preparations. The main source of this impurity are sulphuric acid and the lead apparatus. The I.P. and U.S.P. method is based on the reaction between lead and diathizone (diphenylthiocarbazone).
In chloroform solution dithizone extracts lead from an alkaline aqueous solution as lead dithizone which has red colour in chloroform solution. Since dithizone itself imparts a grÿn colour in chloroform, the resultant colour of dithizone and lead dithizone is violet.
The colour produced by a given amount of the sample is compared with that pro uce y a volume of a standard soluHon of lead. If the colour is intense than that produced by the standard, it contains lead in excess of the prescribed limit.
For the test, the lead present as impurity is separated by extracting an alkaline solution of the substance with dithizone extraction solution which removes all the lead in the form of its complex in chloroform layer.
Limit Test for Arsenic
The presence of arsenic in drugs even in the traces is not desirable because it is toxic and cumulative nature. The Indian Pharmacopeia prescribes the limits for the presence of arsenic
as an impurity in various drugs. For example:- Sodium chloride should not contain arsenic more than 1 parts per million.
Chemically, the arsenic impurity is converted in acidic medium into arsenious acid or. arsenic acid depending upon the valency state of arsenic:

The solution is then reacted with a reducing agent like stannous chloride or sulphurous acid to convert the pentavalent arsenic acid into the trivalent arsenious acid which is converted into gaseous arsenious hydride (arsine gas) with the help of nascent hydroeen produced by the acton of zinc with hydrochloric acid.
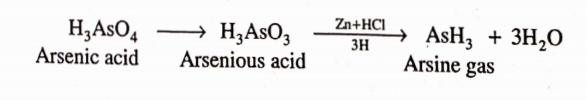
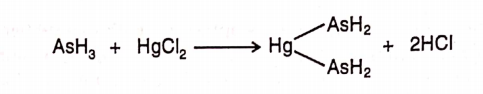
Arsine gas is carried out through the tube with the help of hydrogen to mercuric chloride paper. Reaction of arsine with mercuric chloride produces a yellow coloured stain. The intensity of the colour is dependent on the quantity of arsenic.
Method:
The volume of the prepared sample in a separator is transferred and unless otherwise directed in monograph, 6ml of ammonium citrate solution sp. and 2ml of hydroxylamine hydrochloride solution sp. are added (for the determination of lead in iron salts use 10ml of ammonium citrate solution sp.).
Two drops of phenol red solution are added and the solution made just alkaline (red in colour) by the addition of strong ammonia solution. The solution is cooled, if necessary and 2ml of potassium cyanide solution sp. are added.
Immediately the solution is extracted with several quantities, each of 5ml of dithiazone extraction solution, draining of each extract into another separating funnel, untill the dithiazone extraction solution retains its green colour.
The combined dithizone solutions are shaken for 30 seconds with 30ml of a 1% w/v solution of nitric acid and the chloroform layer discarded.
To the acid solution exactly 5ml of standard dithiazone solution and 4ml of ammonia cyanide solution sp. are added and shaken for 30 seconds, the color of the chloroform layer is of no deeper shade of violet than that of a control made with a volume of dilute standard lead solution equivalent to the amount of lead permitted in the sample under examination.
All reagents used for the test should have as low a content of lead as practicable. All reagent solutions should be stored in containers of borosilicate glass. Glassware should be rinsed throughly with warm dilute nitric acid, followed by water.
Apparatus:
The apparatus consists of a 100ml bottle or conical flask closed with a rubber or ground glass stopper through which passes a glass tube (about 20cm x 5mm). The lower part of the tube is drawn to an internal diameter of 1.0mm and 15mm from its tip is a lateral orifice 2 to 3mm in diameter.
When the tube is in position in the stopper in the lateral orifice should be at least 3mm below the lower surface of the stopper. The upper end of the tube has a perfectly flat surface atupper right angle to the axis of the tube.
A second glass tube of the same internal diameter and 30mm long, with a similar flat surface, is placed in contact with the first and is the lower tube insert 50 to 60mg of lead acetate cotton, loosely packed or a small plug of cotton and a rolled piece of lead acetate paper weighing 50 to 60mg.
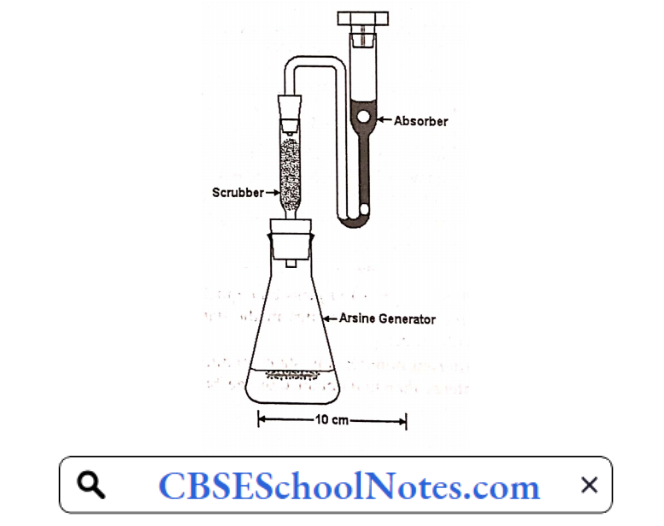
Between the flat surface of the tubes place a disc or a small square of mercuric chloride paper large enough to cover the orifice of the tube (15mm x 15mm). The purpose of lead acetate cotton is to trap any hydrogen sulphide (H2S) gas which would otherwise interfere with this test as it also give some stain with mercuric chloride paper.
The tube is fitted at its upper end with two rubber bungs as shown in figure. A piece of dry mercuricpaper is placed flat on the top of the bung and the other bung is place over it and secured by means of clips in such a manner that the borings of the two bung meet to form a true tube of the same diameter (6.5mm) interrupted by a diaphragm of mercuric chloride paper.
Separate apparatus is used for the test and the standard.
Notes:
- The test is a modification of the Gutzeit test and is, therefore called modified Gutzeit test.
- All the reagents which are used for this test should have a low content of arsenic as possible free from arsenic impurity.
- Potassium iodide is used because it helps in the reduction of pentavalent arsenic acid into trivalent arsenic acid.
- Granulated zinc is used instead of ordinary zinc because evolution of nascent hydrogen is steady and prolonged with granulated zinc.
- The arsenic impurity is expressed in terms of ppm (parts per million). One ppm is lmg in 1kg.
Limit test for Arsenic in Ammonium Chloride:
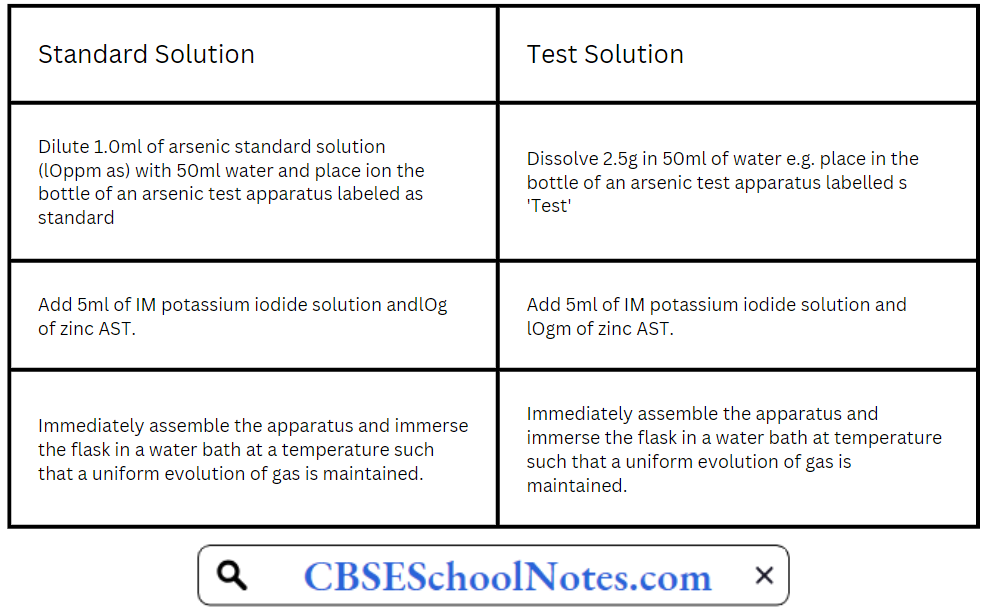
Place the prepared glass tube quickly in position in both the cases and allow the action to proceed for 40 minutes in the dark . compare the yellow stain in the two, in day light without delay.
After 40 minutes any stain produced on the mercuric chloride paper in test is not more intense than that obtained by the ‘Standard’.
Modified procedures for certain limits tests
1. Chloride and Sulphate in Potassium Permanganate
Principle : When limit test of chloride and sulphate are done in usually way, it will be very difficult to make any observation, since the sample itself (Potassium permanganate)
is highly coloured. So the presence of potassium permanganate can be reduced by alcohol. This process is known as PRETREATMENT.
The sample is dissolved in water and heated on a water bath. Alcohol is added. It is filtered to remove the precipitate manganese dioxide. The filterate is colourless & can be used for performing the limit test for chloride and sulphate in the usual way.
Procedure: Dissolve 1.5gm of the sample which is accurately weighed, in 50ml of distilled water. Heat the solution on a water bath and add gradually 6ml of ethanol 95%. Further cool it and dilute to 60ml with distilled water and filter. The filterate (Solution A) is colourless.
For Limit Test for Chlorides
Take 40 ml of solution A and do the limit test for chloride.
For Limit for Sulphate
Take 10 ml of solution A and do the limit test for sulphate.
2. Chloride and Suijp hate in Sodium Bicarbonate
Principle: In the limit test of chloride and sulphate in sodium bicarbonate, the pretreatment consists of neutralising the sodium bicarbonate with an app. Mineral acid and using the neutralised solution for the particular limit test. In the case of limit test for chloride, the sample is dissolved in distilled water & neutralised with nitric acid.
![]()
In the case of limit test for sulphate, the sample is suspended in distilled water and neutralised with HC1.
![]()
In both cases, the solution should be stirred well and the effervescence should be allowed to subside.
Procedure :
For Limit Test of Chloride
Weigh accurately 1.25gm of the sample and dissolve it in 15ml of distilled water. Add 2ml of cone. Nitric acid, then apply the limit test for chloride to this solution
For Limit Test of Sulphate
Weight accurately about lgm of the sample and suspend it in 10ml of distilled water. Neutralise the solution with cone. Hydrochloric acid adding it gradually till the effervescence ceases out. Dilute to 15 ml with distilled water. Do the limit test for sulphate with this solution.
Types Of Impurities In Pharmaceutical Substances Very short answer questions
Question 1. Why only distilled water or purified water is used in performing limit tests?
Answer. In performing limit tests only distilled water or purified water is used because ordinary tap water contains number of ions to validated the test.
Question 2. Why nitric acid is used in performing limit test for chloride?
Answer. Nitric acid is added to prevent precipitation of add radicals such as phosphate, sulphate etc. with silver nitrate because in the presence of nitric add other predpitates are not produced and only chlorides get predpitated.
Question 3. Why HC1 is used in the limit test of sulphate?
Answer. To remove the impurities ofsulphate.
Question 4. Why Barium chloride is used in the limit test of sulphate?
Answer. Barium chloride reacts with a sulphate to produce barium sulphate which can be seen as a white predpitable e.g. therefore can be identified.
Question 5. What is the reagent used in preparingbarium sulphate?
Answer. Sulf + Barium.
Question 6. What do you understand by the term ppm?
Answer. ppm is parts per million. One ppm is lmg in 1kg.
Question 7. Why granulated zinc is used instead of ordinary zinc?
Answer. Granulated zinc is used instead of ordinary zinc because evolution of nascent hydrogen is steady and prolonged with granulated zinc.
Question 8. What do you understand by the term limit tests?
Answer. Limit test is defined as quantitative or semi quantitative test designed to identify and control small quantity of impurity which is likely to be present in the substance.
Question 9. Is there any special apparatus or container required for carrying out limit test?
Answer. Yes Nessler cylinders are required for performing the limit test
Types Of Impurities In Pharmaceutical Substances Fill in the blanks
1. Limit test are qualitative e.g. ………………..test to identify and control small quantities of impurities.
Answer Quantitative
2. Limit test for chloride has been based upon reaction between……… and ……….to obtain silver chloride.
Answer AgN03/Soluble chloride
3. Limit test for sulphate has been based upon the ppt of sulphate with……… in presence of…….
Answer Barium chloride/Hydrochloride
4, Limit test for iron ferrous thioglycolate has stable pink to reddish purple colour in……… medium.
Answer Thioglycolic acid
5. Limit test of iron is based upon reaction of Fe with…………. ammonium citrate
Answer Ferrous mercaptoacetate
6. Limit test for iron purple colour is due to the formation of…….
Answer Alkaline
7. The limit test for arsenic is based upon……..
Answer Guizet test
8. The limit test for arsenic,……… is convertedinto arsenous acid/arsine gas.
Answer Arsine
9. The function of granulated zinc in limit test for arsenic is……
Answer Slow and prolonged evolution of nascent H2 gas.
10. In limit test for heavy metals…… is used for clear colorless/turbid colored solution.
Answer Method A
11. In limit test the change in……..is compared with fixed standard in the pharmacopeia.
Answer Colour, turbidity
12. If the test solution colour, turbidity or opalescence is less than that of the standard solution it……………….the limit test.
Answer Passes or fails