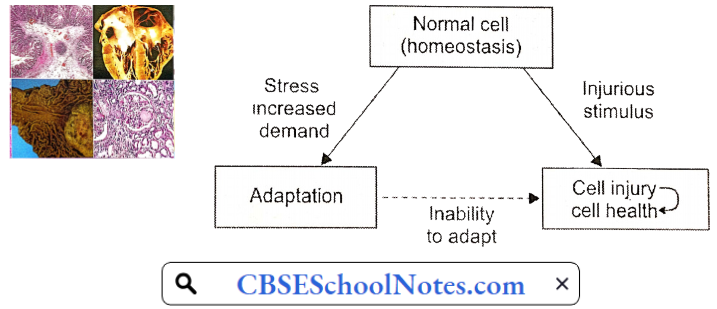
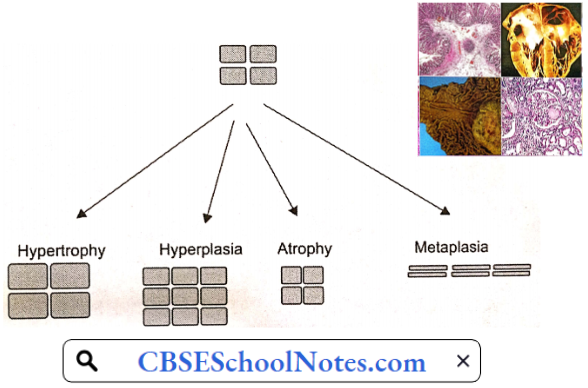
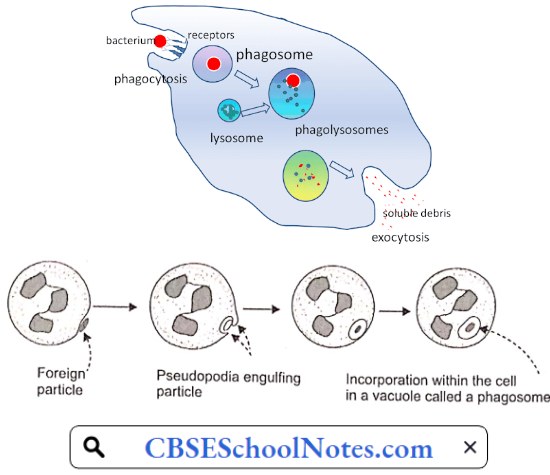
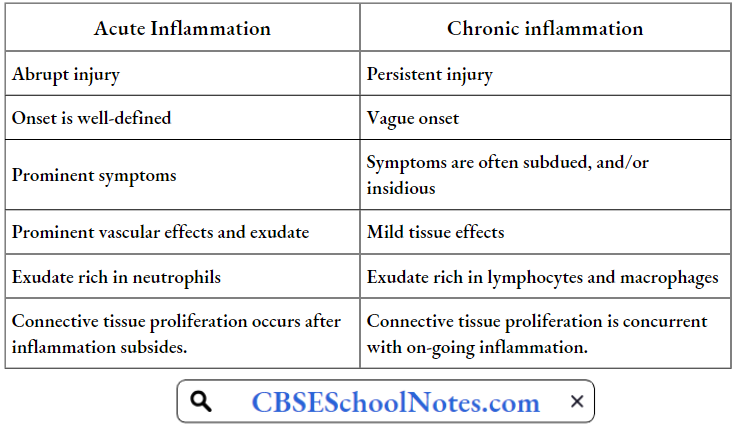
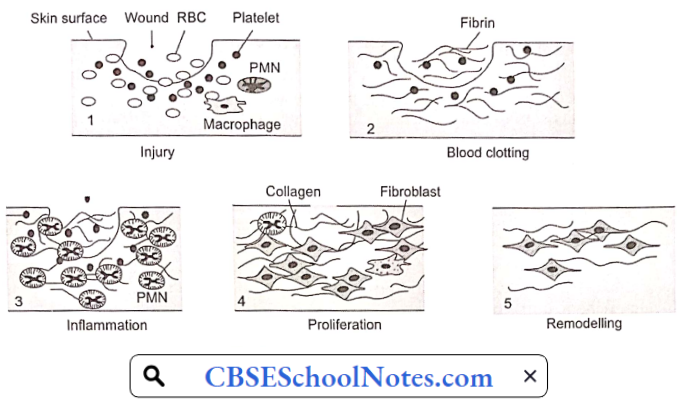
Disorder Of the Cardiovascular System
Hypertension
Hypertension: Hypertension is a serious medical condition that significantly increases the risks of heart, brain, kidney, and other diseases. According to WHO, in 2015, worldwide 25% of males and 20% of females were suffering from hypertension.
Of these, only 20% of patients had BP under control. In India about 33% urban and 25% rural population is hypertensive. More worrying is the report that only one-tenth of rural and one-fifth of urban Indian hypertensive population have their BP under control. Hypertension is directly responsible for 57% of all stroke deaths and 24% of all coronary heart disease deaths in India.
Hypertension Definition: Hypertension is defined as arterial blood pressure greater than 140 mmHg and/or diastolic pressure greater than 90 mmHg. A systolic pressure of 120-140 mmHg or diastolic pressure of 80-90 mmHg is known as prehypertension.
Read and Learn More Pathophysiology
In a vast majority of cases of hypertension, no definite cause can be detected. Such patients are said to suffer from “essential hypertension.” In a small percentage of patients with hypertension, a definite cause can be found, such as kidney disease or/and endocrine disorder. Such cases are said to suffer from secondary hypertension.
Hypertension Symptoms: In most of the cases of high blood pressure, there are no symptoms till the complications occur. When blood pressure is very high, severe headaches may be reported.
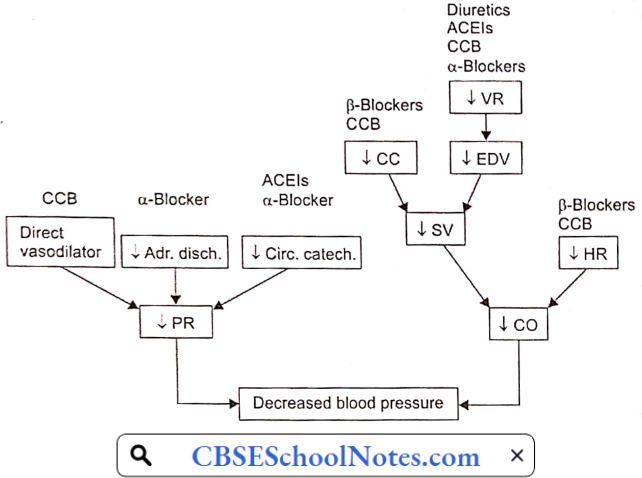
Aetiology And Pathogenesis Of Essential Hypertension: Blood pressure = Cardiac output x Peripheral resistance = CO x PR
Regardless of the origin of hypertension, the actual increase in arterial blood pressure is caused by either an increase in peripheral vascular resistance (PR) or an increase in cardiac output (CO). The former is determined by the vascular tone (i.e. state of constriction) of systemic resistance vessels, whereas the latter is determined by heart rate and stroke volume.
In later stages of hypertension, only peripheral resistance is found to be increased; CO is normal. However, as discussed below, in the early stages of hypertension, many individuals have increased sympathetic activity leading to increased CO.
- Genetic Predisposition: Epidemiological studies have shown the importance of genetic predisposition in the development of essential hypertension. If a family history of hypertension is present, the subject has a 3-4 fold greater chance of developing hypertension, at an age earlier than the general population.
- Although genetics appears to contribute to essential hypertension, the exact mechanism has not been established. Genetic factors interact with environmental factors such as high salt intake, male sex, smoking, obesity, stress and physical inactivity, etc.
- Sympathetic Overactivity: There is evidence for a widespread autonomic abnormality in the early phases of hypertension. Overwhelming and excessive sympathetic activity is consistently present in some patients since their childhood.
- In the early stages of hypertension, blood pressure is elevated when recorded in a doctor’s clinic, but found to be normal when recorded at home. Such cases are said to suffer from “white coat hypertension”. Earlier, it was believed that persons with “white coat hypertension” do not develop established hypertension. There is no support for such an assertion; in fact, such patients are at a high risk of future accelerated hypertension.
- The hallmark of sympathetic over-activity in these patients is the so-called hyperkinetic state that is best characterized by borderline elevation of blood pressure, a fast heart rate, and an increased cardiac output even at rest.
- Both the hyperkinetic state and sympathetic overactivity are less readily recognizable later in the course of hypertension. A large proportion of previously hyperkinetic patients later develop established hypertension.
- It is not clear how from a fast heart rate /high cardiac output form of borderline hypertension is transformed later into the normal cardiac output/high vascular resistance profile that is characteristic of established hypertension.
- Role Of Sodium Intake: Essential hypertension is seen primarily in societies with an average sodium intake above 100 mEq/ day (2.3 g sodium); it is rare in societies with average sodium intakes of less than 50 mEq/day (1.2 g sodium). These epidemiological observations led to the suggestion that the development of hypertension requires a threshold level of sodium intake. This factor effect appears to be independent of other risk factors for hypertension, such as obesity.
- Vascular Hyper-reactivity: Hypertensive patients manifest greater vasoconstrictive response to infused norepinephrine or immersion of one hand in ice-cold water (cold pressor test) than normal individuals.
- Greater vasoconstrictive response to norepinephrine has also been demonstrated in normotensive offspring of hypertensive patients as compared to controls with no family history of hypertension. It suggests that vascular hyperreactivity may be genetic in origin.
- Renin-Angiotensin-Aldosterone-System (RAAS): In patients with essential hypertension, about 15% have mildly elevated plasma renin activity. In another 60% of hypertensives, plasma renin activity is “within the normal range’ but it may be inappropriate in the presence of elevated blood pressure.
- Less than 25% patients of with essential hypertension have subnormal plasma renin activity. Moreover, favorable therapeutic response to RAAS blockers suggests that a renin-dependent mechanism may be involved in tire pathogenesis in about 70% of cases of essential hypertension.
- The fundamental cause of elevated renin activity in such cases is not yet clear. It could be due to chronic sympathetic overactivity. This possibility is supported by the reports that administration of β-blockers in cases with essential hypertension leads to a decrease in plasma renin activity paralleled by a decrease in arterial blood pressure.
- Endothelial dysfunction: Due to its position between the bloodstream and vascular smooth muscle, endothelial dysfunction could either be a consequence or a causative factor in essential hypertension. In recent years, considerable evidence has suggested that changes in vascular endothelial function may cause an increase in vascular tone.
- For example, in hypertensive patients, the vascular endothelium produces less nitric oxide (intrinsic vasodilator). Moreover, the vascular smooth muscle is less sensitive to the actions of this powerful vasodilator. There may also be an increase in endothelin (a vasoconstrictor) production by the endothelial cells, which can enhance vasoconstrictor tone.
Complications Of Untreated Essential Hypertension
- Atherosclerosis: Many of the complications of hypertension are related to the effects of sustained elevations of blood pressure on vasculature and heart. Atherosclerosis is commonly associated with and is accelerated by long-standing hypertension.
- Most of the adverse outcomes in hypertension are associated with thrombosis rather than bleeding. Atherosclerosis predisposes the hypertensive patient to coronary thrombosis and cerebral stroke. Cerebral strokes are more often due to thrombosis rather than hemorrhage in the cerebral vessels.
- The excess morbidity and mortality related to hypertension are progressive over the whole range of systolic and diastolic blood pressures and are not limited to high values only. However, target-organ damage varies markedly between individuals with similar levels of hypertension. Atherosclerosis may also result in aortic aneurysm or peripheral arterial disease.
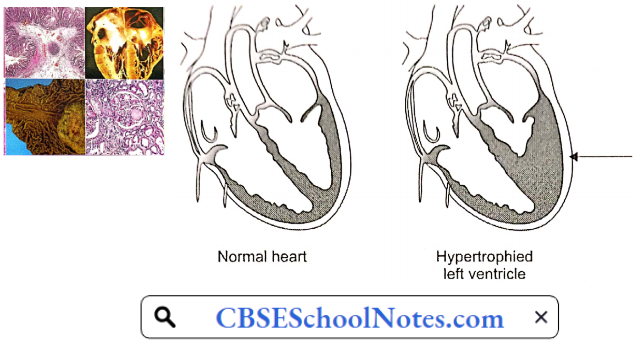
- Hypertensive Cardiomyopathy: A sustained increase in blood pressure (afterload) results in hypertrophy and subsequent dilatation of the left ventricle. Electrocardiographic evidence of left ventricular hypertrophy is found in up to 15% of persons with chronic hypertension. Left ventricular hypertrophy may cause or facilitate many cardiac complications of hypertension, including myocardial ischemia, congestive heart failure, and ventricular arrhythmias.
- Cerebrovascular Complications: Hypertension is an important risk factor for cerebral stroke. Approximately 85% of strokes are due to thrombosis and the remainder are due to hemorrhage in cerebral blood vessels.
- The term hypertensive encephalopathy is used to describe a group of symptoms and signs that sometimes follow a sudden and sustained rise in blood pressure. The symptoms are characterized by a severe headache, restlessness, impaired judgment, and memory, confusion, somnolence, and stupor. If the condition is not treated, these neurological symptoms may worsen and ultimately turn into a coma.
- Cerebral encephalopathy seems to result from a failure of autoregulation of cerebral blood flow. The autoregulation seems to fail when hypertension becomes excessive.
- Retinopathy: The primary response of the retinal arterioles to systemic hypertension is vasoconstriction. However, sustained hypertension leads to disruption of the blood—retinal barrier, increased vascular permeability, and secondary arteriolosclerosis. Loss of vision may occur.
- Renal Complications: Renal failure is one of the important complications of chronic hypertension. Sustained elevation of blood pressure damages renal microvasculature. Renal damage itself is a cause of hypertension (see secondary hypertension below), starting a vicious cycle.
- Sexual Dysfunction: Sexual dysfunction is more common and more severe in men with hypertension than it is in the general population. Hypertension is itself the major cause of erectile dysfunction. Experimental studies indicate that essential hypertension results in structural and functional changes in penile vasculature.
- Cavernous vessels are affected by chronic elevation of arterial blood pressure in the same fashion as other blood vessels. Marked hypertrophy in the smooth muscle of cavernous vessels, increased smooth muscle layer in cavernous space, and increased extracellular matrix (collagen) explain the pathophysiological mechanism of erectile dysfunction in essential hypertension.
Pathophysiological Basis Of Treatment Of Essential Hypertension
Non-Pharmacological Measures
- Reduction or elimination of factors such as stress, smoking, obesity
- Regular aerobic exercise
- Restriction of dietary calories, salt, cholesterol, and saturated fats
Pharmacological Measures: A variety of drugs are being used in the treatment of essential hypertension. They reduce cardiac output, peripheral resistance, or both.
Secondary Hypertension: Secondary hypertension is defined as hypertension that is caused by an underlying well-defined primary cause. It is much less common than essential hypertension, affecting only 5% of hypertensive patients. Some of the causes of secondary hypertension are treatable.
Secondary Hypertension Symptoms: Like primary hypertension, secondary hypertension usually has no specific signs or symptoms, even if blood pressure has reached dangerously high levels.
Secondary Hypertension Causes
- Chronic kidney disease (chronic glomerulonephritis)
- Renal artery stenosis
- Cushing syndrome (increased secretion of cortisol by a tumor of the adrenal cortex).
- Aldosteronism (increased secretion of aldosterone by a tumor of the adrenal cortex).
- Pheochromocytoma (increased secretion of norepinephrine and epinephrine by a tumor of the adrenal medulla).
- Coarctation of the aorta: There is congenital narrowing (coarctation) in the thoracic aorta. Hypertension is recorded in the upper part of the body only.
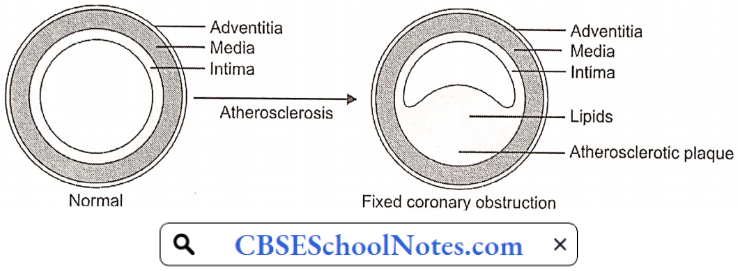
Atherosclerosis
Arteriosclerosis is the thickening, hardening, and loss of elasticity of the walls of arteries. This process gradually restricts the blood flow to one’s organs and tissues and can lead to severe organ damage. Atherosclerosis, which is a specific form of arteriosclerosis, is caused by the build-up of fatty acid-cholesterol plaques in the artery walls.
Atherosclerosis: Atherosclerosis develops primarily in large elastic arteries, for example, the aorta and carotid arteries, or large or medium-sized muscular arteries such as coronary, cerebral, renal, and popliteal arteries.
Atherosclerosis can lead to serious complications such as coronary artery disease, cerebral stroke, and peripheral arterial disease (gangrene of feet or legs). Risk factors associated with atherosclerosis, include:
- Elevated blood cholesterol and triglyceride levels
- High blood pressure
- Obesity
- Smoking
- Physical inactivity
- Diabetes mellitus
Atherosclerosis Symptoms: Atherosclerosis develops gradually. Mild atherosclerosis usually does not have any symptoms. Symptoms develop only when the narrowing of the artery is so severe that an adequate amount of blood does not reach the tissues or organs. At that time symptoms depend on the artery affected.
- Coronary Artery: Symptoms of angina or myocardial infarction
- Cerebral Artery: Symptoms of cerebral stroke.
- Renal Artery: Development of hypertension or symptoms of renal failure.
- Popliteal Artery: Pain in the legs while walking (claudication)
Pathogenesis Of Atherosclerosis: Aetiological factors named above induce hypercholesterolemia, which disturbs vascular homeostasis, including a decrease in nitrous oxide bioactivity, an increase in superoxide production, an increase in adhesion molecules, and attenuation of endothelium-dependent vasodilatation.
The earliest pathologic lesion of atherosclerosis is the fatty streak. The fatty streak is the result of focal accumulation of serum lipoproteins within the intima of the vessel wall. Gradually, the fatty streak progresses to form a fibrous plaque.
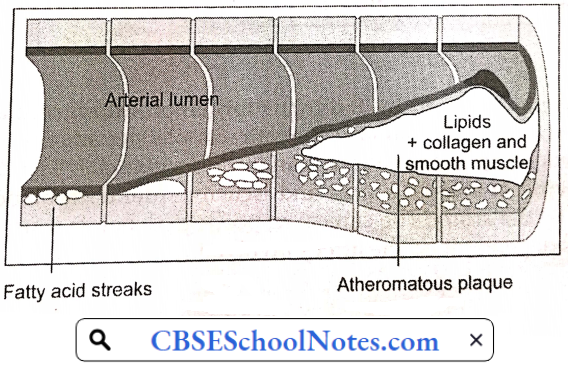
Circulating monocytes infiltrate the intima of the vessel wall. The combination of diabetes and hypertension appears to have an additive effect on monocyte adhesion. These tissue macrophages act as scavenger cells, taking up LDL cholesterol and forming the characteristic foam cells of early atherosclerosis.
These activated macrophages produce numerous factors that are injurious to the endothelium. Atherosclerotic plaque is the result of progressive lipid accumulation along with migration and proliferation of smooth muscle cells. Growth of the fibrous plaque results in progressive luminal narrowing.
Microscopy reveals lipid-laden macrophages, T-lymphocytes, and smooth muscle cells in varying proportions. Atheromatous plaques convert the smooth lining of the tunica intima of the blood vessels to a roughened surface prone to thrombosis.

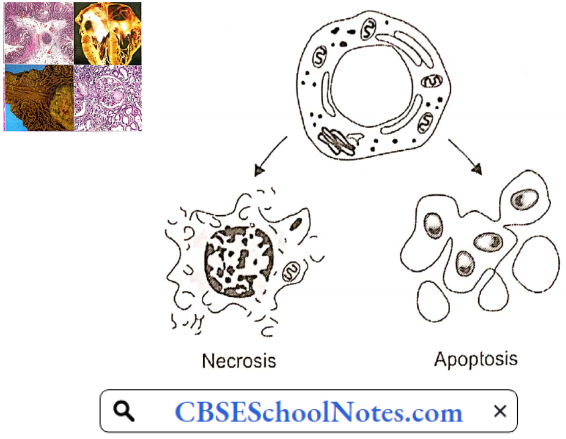
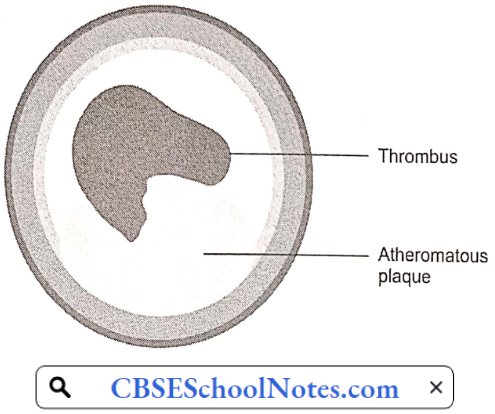
Moreover, developing atherosclerotic plaques are prone to necrosis. Loss of the overlying endothelium or rupture of the protective fibrous cap may result in exposure of the thrombogenic contents of the core of the plaque to the circulating blood.
This exposure constitutes an advanced or complicated lesion. A plaque rupture may result in thrombus formation leading to partial or complete occlusion of the blood vessel.
Complications Of Atherosclerosis
Thrombosis: Rupture of plaque is followed by thrombus formation (intravascular clotting). The rough endothelial lining of a blood vessel attracts platelet adhesion and activation. Activated platelets result in the formation of an intravascular clot.
The thrombus results in critical narrowing of the arterial lumen and ischemia (deficient blood supply) in the tissues supplied by the artery. The clinical response to ischemia caused by obstructive atherosclerosis is dependent on the artery involved:
Coronary Artery Disease
Cardiovascular diseases are the number 1 cause of death globally. An estimated 17.9 million people died from CVDs in 2016, representing 31% of all global deaths. Of these deaths, 85% are due to heart attack and stroke.
In India, studies have reported an increasing prevalence of coronary artery disease over the last 60 years, from 1% to 9-10% in urban populations and <1% to 4-6% in rural populations.
Myocardial Oxygen Supply
- The myocardial oxygen supply depends on:
- The oxygen content of the arterial blood, and
The rate of coronary blood flow. The oxygen content of arterial blood may be decreased because of decreased hemoglobin concentration or because of poor systemic blood oxygenation (hypoxic hypoxia). Thus, angina may be a presenting feature of a patient with severe anemia or lung disease.
- In the absence of anemia or lung disease, oxygen supply to the heart is determined by the rate of coronary blood flow.
- In most other organs, because of the greater pressure head, blood flow is greater during systole than in diastole of the heart. However, in the case of the myocardium, the reverse is true. The coronary arteries that run on the surface of the heart are called epicardial coronary arteries.
- Branches of epicardial arteries that run into and supply blood to the myocardium are called subendocardial coronary vessels. During systole, myocardial contraction has a strangulating effect on the blood vessels passing through the cardiac muscle fibers. Because of this, blood flow in the subendocardial vessels stops.
- As a result, most myocardial perfusion occurs during diastole when the subendocardial coronary vessels are patent because of the absence of extramural pressure.
- Although coronary vessels are supplied with sympathetic and parasympathetic nerve fibers, coronary vascular resistance is chiefly determined by intrinsic metabolic factors rather than neural control.
- Local vasodilator metabolites such as adenosine (chiefly) and other products of anoxic metabolism (lactate, H+, certain prostaglandins) regulate coronary blood flow by a direct action on vascular smooth muscle. During exercise, greater release of local vasodilator metabolites assures greater blood flow in the coronary arteries.
- Atherosclerotic narrowing of coronary arteries produces its effects mainly by hypo-perfusion (decreased blood supply) of the myocardium. The effect may range from angina to myocardial infarction.
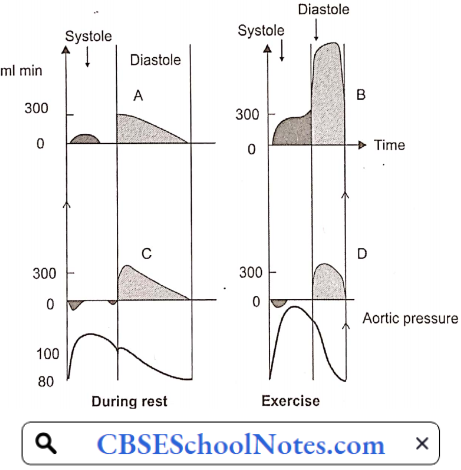
Angina Pectoris: In normal individuals during exercise, by the local metabolite control, coronary arteriolar resistance decreases in proportion to the increase in O2 demand of the myocardium. Thus, coronary blood flow increases in proportion to the oxygen demand of the myocardium.
- When atherosclerotic narrowing is greater than 60-70%, coronary blood flow cannot increase during exercise in spite of the presence of vasodilator metabolites. Therefore, myocardial ischemia results; which is commonly intermittent (only during exertion).
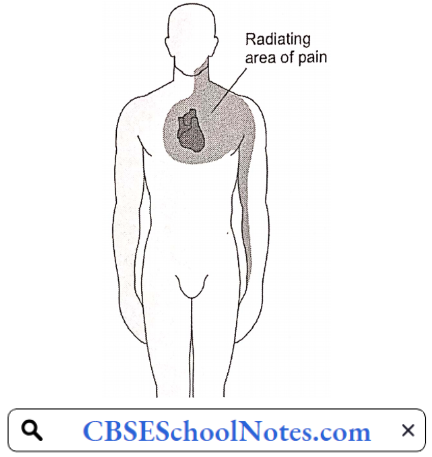
- Anginal pain is characterized by the fact that it occurs only at times of increased myocardial oxygen demand such as exertion or emotional excitement, but subsides by rest. Such a condition is known as angina. In angina, pain may be localized to the substernum or referred to the left arm, neck, or jaw.
Angina Pectoris Symptoms: Angina symptoms include
- Pain or discomfort can spread to the chest, jaw, shoulders, arms (mostly the left arm), and back.
- Chest tightness, burning, heaviness, feeling of squeezing or not being able to breathe.
- Angina will sometimes cause dizziness, paleness, and weakness.
The Mechanisms Of Cardiac Pain: It is presumed that pain of angina pectoris results from the release of anoxic metabolites (adenosine, bradykinin) by the myocardium. These metabolites excite the sensory ends of the sympathetic and vagal afferent fibers supplying the heart.
Within the spinal cord, cardiac sympathetic afferent impulses may converge with impulses from somatic thoracic structures, which may be the basis for referred cardiac pain, for example, to the left arm.
Pathophysiologic Basis Of Treatment Of Angina: For the immediate relief of angina pain, the patient is advised to take a sublingual tablet of nitroglycerin.
- Nitroglycerin is converted to a powerful vasodilator nitric oxide (NO) in the body. It may produce some dilation of coronary arteries, but the major action is venous vasodilation. Vasodilation causes the pooling of blood within the venous system, reducing preload to the heart.
- This causes a decrease in cardiac work, and cardiac oxygen demand and hence relieves angina pain.
Myocardial Infarction
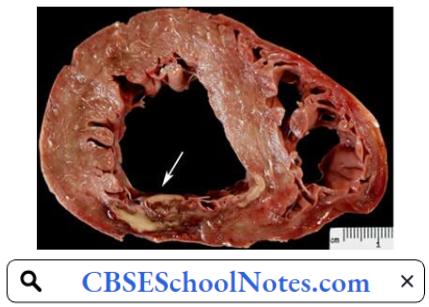
When the myocardial ischemia progresses to a degree that irreversible necrosis of a part of the myocardium occurs, an acute myocardial infarction (MI) is said to have occurred. An acute MI almost always results from an acute thrombotic obstruction of an atherosclerotic coronary artery.
Myocardial Infarction Symptoms
- Chest pain or discomfort, possibly described as pressure, squeezing, burning, or fullness
- Pain in the left arm, neck, jaw, shoulder, or back accompanying chest pain
- Nausea
- Fatigue
- Shortness of breath
- Sweating
- Dizziness
In acute MI, the pain has the same characteristics as angina, but it is far more severe, lasts longer, may radiate more widely, and is not relieved by rest or nitroglycerin. Pain may be due to the accumulation of anoxic metabolites as well as products of tissue necrosis. The pain is accompanied by greater psychogenic effects, i.e. feeling of impending death.
Myocardial Infarction Pathogenesis: Acute myocardial infarction (MI) indicates irreversible myocardial injury resulting in necrosis of a significant portion of myocardium (generally >1 cm). Myocardial infarction is usually due to thrombotic occlusion of a coronary vessel caused by rupture of a vulnerable plaque.
Ischemia induces profound metabolic and ionic perturbations in the affected myocardium. Prolonged myocardial ischemia results in ischemic necrosis of the myocardium. The adult mammalian heart has negligible regenerative capacity, thus the infarcted myocardium heals through the formation of a scar.
Infarct healing is intertwined with the geometric remodeling of the chamber, characterized by dilation, hypertrophy of viable segments, and progressive dysfunction. Cell membrane damage in acute MI leads to the release of certain intracellular enzymes. An increase in their plasma levels is used as diagnostic evidence of myocardial infarction.
Myocardial Infarction Risk Factors
- Age: Men age 45 or older and women age 55 or older are more likely to have a heart attack than are younger men and women.
- Tobacco: This includes smoking and long-term exposure to second-hand smoke.
- High Blood Pressure: Over time, high blood pressure can damage coronary arteries.
- High Blood Cholesterol Or Triglyceride Levels: A high level of low-density lipoprotein (LDL) cholesterol (the ‘bad’ cholesterol) is most likely to narrow arteries. A high level of triglycerides, a type of blood fat related to your diet, also increases your risk of heart attack.
- Obesity: Obesity is associated with high blood cholesterol levels, high triglyceride levels, high blood pressure, and diabetes.
- Diabetes.
- Family history of heart attack
- Lack of physical activity
- Stress.
Myocardial Infarction Diagnosis
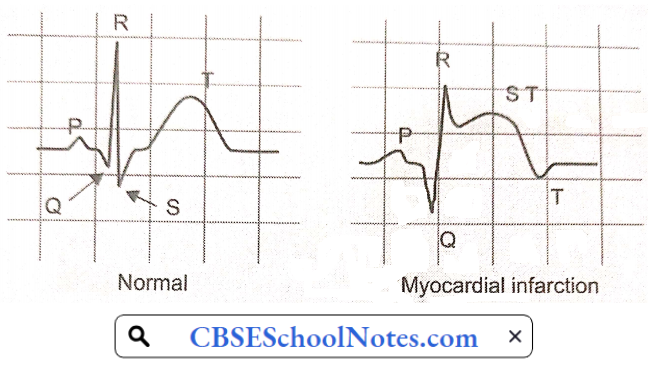
- Electrocardiogram: An electrocardiogram (ECG) is a recording of the electrical activity of the heart. Abnormalities in electrical activity usually occur with heart attacks and can identify the areas of the heart muscle that are deprived of oxygen and/or areas of muscle that have died.
- In a patient with typical symptoms of heart attack (such as crushing chest pain) and characteristic changes of heart attack on the ECG, a secure diagnosis of heart attack can be made quickly in the emergency room and treatment can be started immediately.
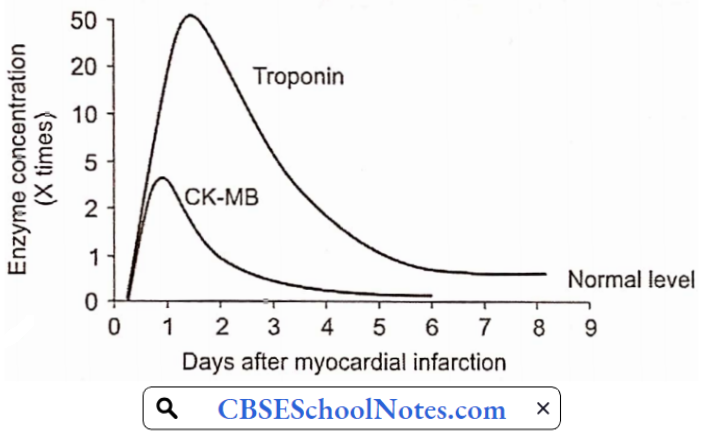
- Blood Tests: Cardiac enzymes are proteins that are released into the blood by dying heart muscles. These cardiac enzymes are creatine kinase (CK-MB), and troponin, and their levels can be measured in blood.
- These cardiac enzymes typically are elevated in the blood several hours after the onset of a heart attack. Currently, troponin levels are considered the preferred lab tests to use to help diagnose a heart attack, as they are indicators of cardiac muscle injury or death.
A series of blood tests for tire enzymes performed over a 24-hour period are useful not only in confirming the diagnosis of heart attack, but the changes in their levels over time also correlate with the amount of heart muscle that has died.
Complications Of MI: More important complications include
- Cardiogenic shock
- Cardiac arrhythmias (ventricular tachycardia or ventricular fibrillation are life-threatening complications).
- Congestive heart failure
Pathophysiologic Basis Of Treatment: Treatment aims at immediate restoration of blood flow in the artery blocked by a blood clot followed by measures to prevent thrombosis again
- Angioplasty And Stent: Special tubing with an attached deflated balloon is threaded up to the coronary arteries. A stent is a wire mesh tube used to prop open an artery during angioplasty.
- Long-term antiplatelet therapy
Congestive Heart Failure
Congestive heart failure (CHF), or heart failure is defined as an inability of the heart to pump blood at a rate appropriate for the metabolic requirements of the tissues;
- Aetiology: Important causes include:
- Hypertension: Due to high blood pressure, expulsion of blood requires more forceful ventricular contraction during each systole. Over time, the left ventricle initially undergoes hypertrophy and later it dilates leading to heart failure.
- Coronary Artery Disease: Myocardial infarction weakens the myocardium.
- Valvular Disease: Disorders of aortic or mitral valve cause extra burden on the heart. Initially, the heart undergoes hypertrophy and later dilatation.
Congestive Heart Failure Symptoms And Signs
- Exercise intolerance
- Fatigue
- Dyspnoea on effort
- Cyanosis
- Ankle edema
- Distended neck veins
- Liver enlargement (hepatomegaly)
- Spleen enlargement (splenomegaly)
- Oedema feet
- Ascites (fluid in the peritoneal cavity, abdomen)

Pathophysiology Of CHF: When the heart is unable to pump out a sufficient amount of blood, a number of natural compensatory mechanisms are activated so as to improve the cardiac output and maintain normal perfusion of the vital organs. These mechanisms include:
- Frank-Starling mechanism
- Increased adrenergic discharge
- Regional redistribution of cardiac output
- Hormonal mechanisms
As you have learned in physiology lectures, cardiac output can be increased by an increase in end-diastolic volume (EDV) (Starling mechanism) as well as an increase in sympathetic discharge to the heart. Both of these mechanisms are utilized to bring the failing heart to produce normal cardiac output.
The other two compensatory mechanisms help in these primary mechanisms are:
- Frank-Starling Mechanism: By working at greater EDV (Y), the heart can pump out a normal amount of blood. EDV is increased by the fourth compensatory mechanism mentioned above.
- Increased Adrenergic Discharge: In the failing heart, depressed cardiac output is sensed by high-pressure baroreceptors located in the carotid sinus and aortic arch, leading to a reflex increase in adrenergic discharge to the heart and blood vessels.
- In the heart, increased adrenergic discharge improves the cardiac output by increasing the heart rate as well as stroke volume. In the blood vessels, it causes a redistribution of cardiac output described next.
- Redistribution Of Cardiac Output: The redistribution of cardiac output serves as an important compensatory mechanism when cardiac output is reduced. Blood flow is redistributed so that the delivery of oxygen to vital organs, such as the brain and myocardium, is maintained at normal or near-normal levels, while flow to less critical areas, such as the cutaneous and muscular beds and viscera, is reduced. Vasoconstriction mediated by the adrenergic nervous system is largely responsible for this redistribution.
- Hormonal Mechanisms: Decreased cardiac output activates the renin-angiotensin II mechanism. Angiotensin II produces arteriolar constriction and increases thirst (increased water intake). It also increases aldosterone secretion, thereby producing salt and water retention in the kidney. As a result of both these factors, blood volume and thereby EDV is increased.
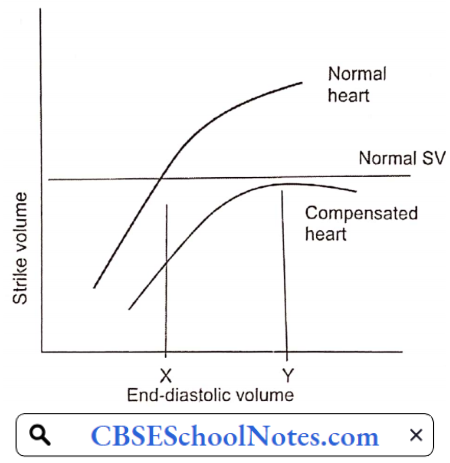
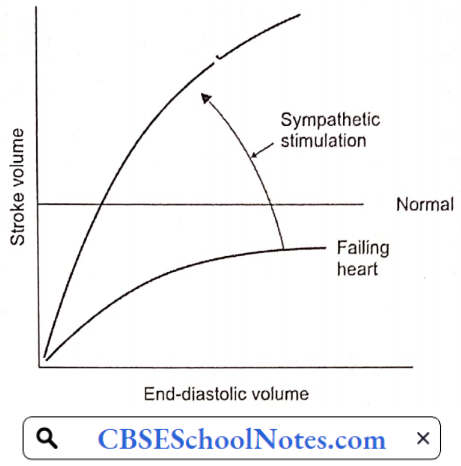
Congestive Heart Failure Decompensation: As the cardiac function gradually deteriorates, the compensatory mechanisms discussed above cannot maintain normal cardiac output. Moreover, excessive increase in EDV increases left and right atrial pressures resulting in congestion in the lungs and in systemic veins. At this point, congestive heart failure is said to have set in.
Pathophysiological Basis of Treatment
- Strengthening The Force Of Contraction Of The Heart: Digitalis.
- Reducing Salt And Water Retention: Diuretics
- Reducing Sympathetic Over-Activity: Beta-blockers